

Clinical and molecular characterization of 40 patients with classic Ehlers-Danlos syndrome: identification of 18 COL5A1 and 2 COL5A2 novel mutations
- PMID: 23587214
- PMCID: PMC3653713
- DOI: 10.1186/1750-1172-8-58
Clinical and molecular characterization of 40 patients with classic Ehlers-Danlos syndrome: identification of 18 COL5A1 and 2 COL5A2 novel mutations
Abstract
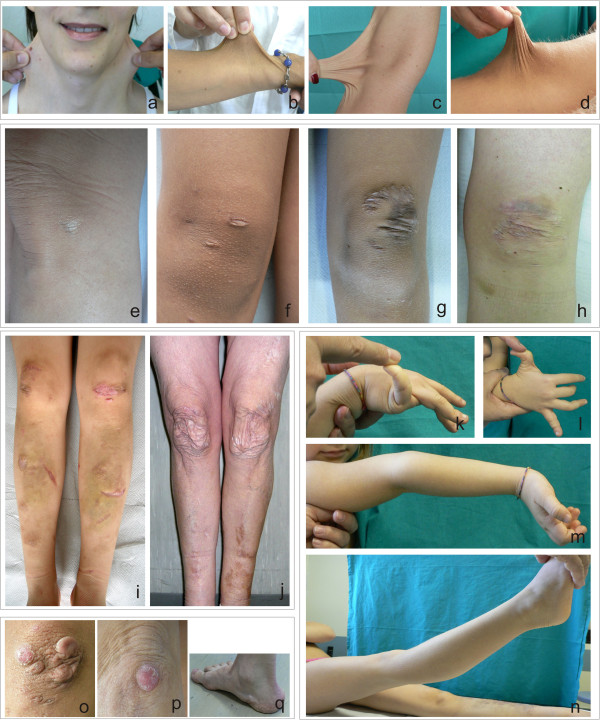
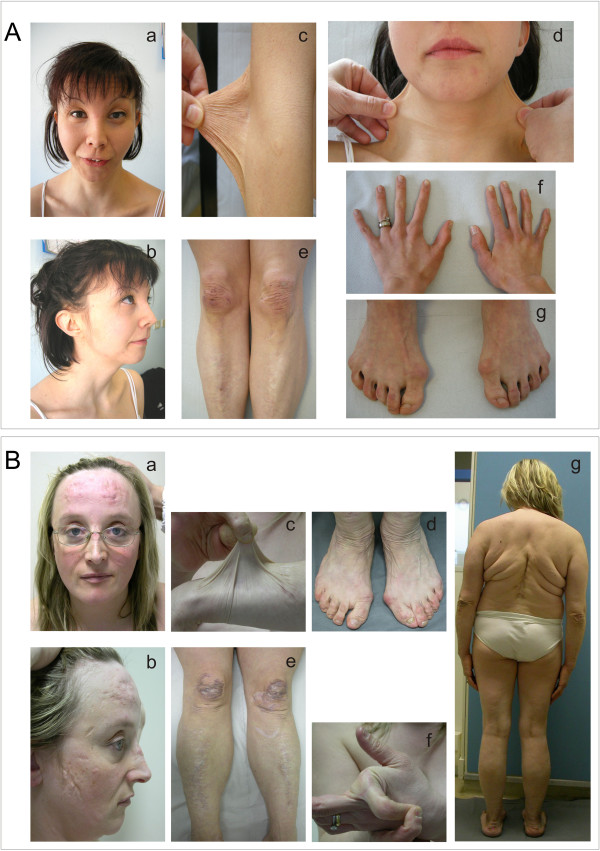
Background: Classic Ehlers-Danlos syndrome (cEDS) is a rare autosomal dominant connective tissue disorder that is primarily characterized by skin hyperextensibility, abnormal wound healing/atrophic scars, and joint hypermobility. A recent study demonstrated that more than 90% of patients who satisfy all of these major criteria harbor a type V collagen (COLLV) defect.
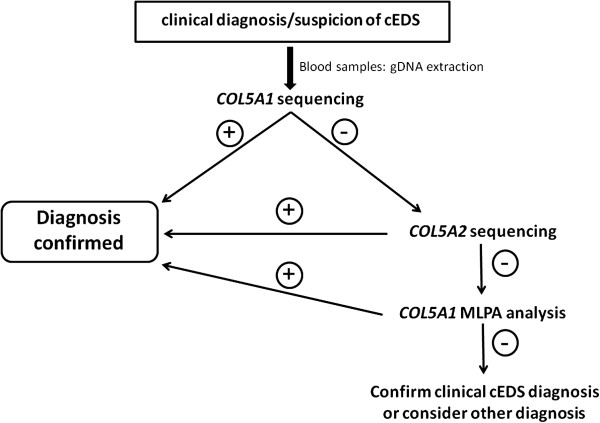
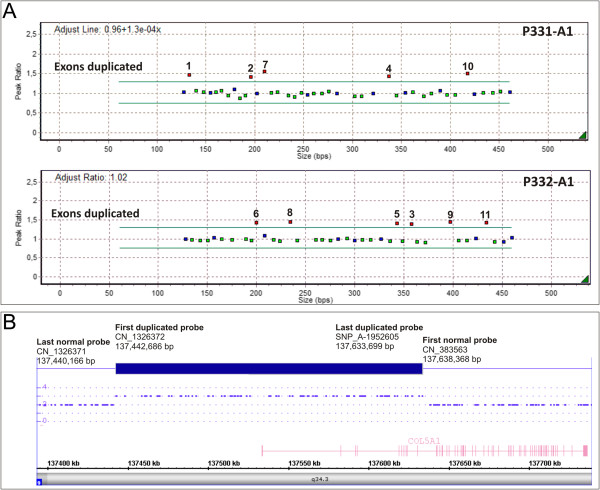
Methods: This cohort included 40 patients with cEDS who were clinically diagnosed according to the Villefranche nosology. The flowchart that was adopted for mutation detection consisted of sequencing the COL5A1 gene and, if no mutation was detected, COL5A2 analysis. In the negative patients the presence of large genomic rearrangements in COL5A1 was investigated using MLPA, and positive results were confirmed via SNP-array analysis.
Results: We report the clinical and molecular characterization of 40 patients from 28 families, consisting of 14 pediatric patients and 26 adults. A family history of cEDS was present in 9 patients. The majority of the patients fulfilled all the major diagnostic criteria for cEDS; atrophic scars were absent in 2 females, skin hyperextensibility was not detected in a male and joint hypermobility was negative in 8 patients (20% of the entire cohort). Wide inter- and intra-familial phenotypic heterogeneity was observed. We identified causal mutations with a detection rate of approximately 93%. In 25/28 probands, COL5A1 or COL5A2 mutations were detected. Twenty-one mutations were in the COL5A1 gene, 18 of which were novel (2 recurrent). Of these, 16 mutations led to nonsense-mediated mRNA decay (NMD) and to COLLV haploinsufficiency and 5 mutations were structural. Two novel COL5A2 splice mutations were detected in patients with the most severe phenotypes. The known p. (Arg312Cys) mutation in the COL1A1 gene was identified in one patient with vascular-like cEDS.
Conclusions: Our findings highlight that the three major criteria for cEDS are useful and sufficient for cEDS clinical diagnosis in the large majority of the patients. The borderline patients for whom these criteria fail can be diagnosed when minor signs of connective tissue diseases and family history are present and when genetic testing reveals a defect in COLLV. Our data also confirm that COL5A1 and COL5A2 are the major, if not the only, genes involved in cEDS.
Figures
Similar articles
-
Molecular insights in the pathogenesis of classical Ehlers-Danlos syndrome from transcriptome-wide expression profiling of patients' skin fibroblasts.PLoS One. 2019 Feb 4;14(2):e0211647. doi: 10.1371/journal.pone.0211647. eCollection 2019. PLoS One. 2019. PMID: 30716086 Free PMC article.
-
Identification of the novel COL5A1 c.3369_3431dup, p.(Glu1124_Gly1144dup) variant in a patient with incomplete classical Ehlers-Danlos syndrome: The importance of phenotype-guided genetic testing.Mol Genet Genomic Med. 2020 Oct;8(10):e1422. doi: 10.1002/mgg3.1422. Epub 2020 Jul 28. Mol Genet Genomic Med. 2020. PMID: 32720758 Free PMC article.
-
Clinical and genetic aspects of Ehlers-Danlos syndrome, classic type.Genet Med. 2010 Oct;12(10):597-605. doi: 10.1097/GIM.0b013e3181eed412. Genet Med. 2010. PMID: 20847697 Review.
-
Delineation of Ehlers-Danlos syndrome phenotype due to the c.934C>T, p.(Arg312Cys) mutation in COL1A1: Report on a three-generation family without cardiovascular events, and literature review.Am J Med Genet A. 2017 Feb;173(2):524-530. doi: 10.1002/ajmg.a.38035. Epub 2016 Nov 7. Am J Med Genet A. 2017. PMID: 28102596 Review.
-
Multisystemic manifestations in a cohort of 75 classical Ehlers-Danlos syndrome patients: natural history and nosological perspectives.Orphanet J Rare Dis. 2020 Jul 31;15(1):197. doi: 10.1186/s13023-020-01470-0. Orphanet J Rare Dis. 2020. PMID: 32736638 Free PMC article.
Cited by
-
Haploinsufficiency of Col5a1 causes intrinsic lung and respiratory changes in a mouse model of classical Ehlers-Danlos syndrome.Physiol Rep. 2022 Apr;10(8):e15275. doi: 10.14814/phy2.15275. Physiol Rep. 2022. PMID: 35439366 Free PMC article.
-
Main and Minor Types of Collagens in the Articular Cartilage: The Role of Collagens in Repair Tissue Evaluation in Chondral Defects.Int J Mol Sci. 2021 Dec 11;22(24):13329. doi: 10.3390/ijms222413329. Int J Mol Sci. 2021. PMID: 34948124 Free PMC article. Review.
-
Molecular insights in the pathogenesis of classical Ehlers-Danlos syndrome from transcriptome-wide expression profiling of patients' skin fibroblasts.PLoS One. 2019 Feb 4;14(2):e0211647. doi: 10.1371/journal.pone.0211647. eCollection 2019. PLoS One. 2019. PMID: 30716086 Free PMC article.
-
Clinical and genetic analysis of classical Ehlers-Danlos syndrome patient caused by synonymous mutation in COL5A2.Mol Genet Genomic Med. 2021 May;9(5):e1632. doi: 10.1002/mgg3.1632. Epub 2021 Apr 8. Mol Genet Genomic Med. 2021. PMID: 33834621 Free PMC article.
-
The Ehlers-Danlos syndromes.Nat Rev Dis Primers. 2020 Jul 30;6(1):64. doi: 10.1038/s41572-020-0194-9. Nat Rev Dis Primers. 2020. PMID: 32732924 Review.
References
-
- Steinmann B, Royce PM, Superti-Furga A. In: Connective tissue and its heritable disorders: molecular genetics and medicals aspects. Royce PM, Steinmann B, editor. New York: Wiley-Liss; 2002. The Ehlers-Danlos syndrome; pp. 351–407.
-
- Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers Danlos Support Group (UK) Am J Med Genet. 1998;77:31–37. doi: 10.1002/(SICI)1096-8628(19980428)77:1<31::AID-AJMG8>3.0.CO;2-O. - DOI - PubMed
-
- Malfait F, Wenstrup R, De Paepe A. In: GeneReviews™ [internet]. Seattle (WA) Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, editor. Seattle: University of Washington; 1993. Ehlers-Danlos syndrome, classic type. updated 2011 Aug 18.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
