

Haploinsufficiency of a spliceosomal GTPase encoded by EFTUD2 causes mandibulofacial dysostosis with microcephaly
- PMID: 22305528
- PMCID: PMC3276671
- DOI: 10.1016/j.ajhg.2011.12.023
Haploinsufficiency of a spliceosomal GTPase encoded by EFTUD2 causes mandibulofacial dysostosis with microcephaly
Abstract
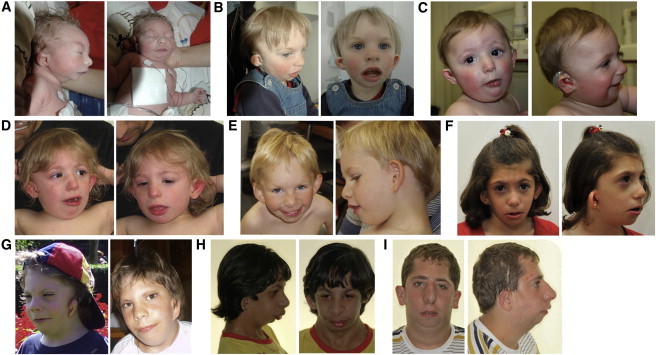
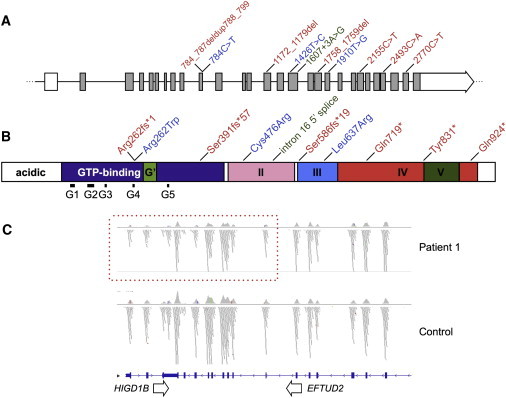
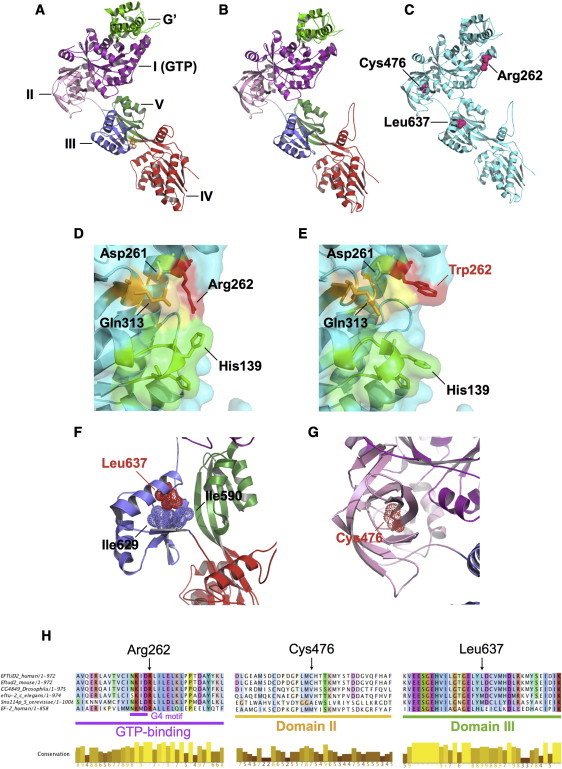
Mandibulofacial dysostosis with microcephaly (MFDM) is a rare sporadic syndrome comprising craniofacial malformations, microcephaly, developmental delay, and a recognizable dysmorphic appearance. Major sequelae, including choanal atresia, sensorineural hearing loss, and cleft palate, each occur in a significant proportion of affected individuals. We present detailed clinical findings in 12 unrelated individuals with MFDM; these 12 individuals compose the largest reported cohort to date. To define the etiology of MFDM, we employed whole-exome sequencing of four unrelated affected individuals and identified heterozygous mutations or deletions of EFTUD2 in all four. Validation studies of eight additional individuals with MFDM demonstrated causative EFTUD2 mutations in all affected individuals tested. A range of EFTUD2-mutation types, including null alleles and frameshifts, is seen in MFDM, consistent with haploinsufficiency; segregation is de novo in all cases assessed to date. U5-116kD, the protein encoded by EFTUD2, is a highly conserved spliceosomal GTPase with a central regulatory role in catalytic splicing and post-splicing-complex disassembly. MFDM is the first multiple-malformation syndrome attributed to a defect of the major spliceosome. Our findings significantly extend the range of reported spliceosomal phenotypes in humans and pave the way for further investigation in related conditions such as Treacher Collins syndrome.
Copyright © 2012 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Mandibulofacial Dysostosis with Microcephaly: Mutation and Database Update.Hum Mutat. 2016 Feb;37(2):148-54. doi: 10.1002/humu.22924. Epub 2015 Nov 19. Hum Mutat. 2016. PMID: 26507355 Free PMC article. Review.
-
Mandibulofacial dysostosis with microcephaly: An expansion of the phenotype via parental survey.Am J Med Genet A. 2021 Feb;185(2):413-423. doi: 10.1002/ajmg.a.61977. Epub 2020 Nov 27. Am J Med Genet A. 2021. PMID: 33247512 Free PMC article.
-
A novel de novo missense mutation in EFTUD2 identified by whole-exome sequencing in mandibulofacial dysostosis with microcephaly.J Clin Lab Anal. 2022 May;36(5):e24440. doi: 10.1002/jcla.24440. Epub 2022 Apr 18. J Clin Lab Anal. 2022. PMID: 35435265 Free PMC article.
-
Clinical and molecular delineation of mandibulofacial dysostosis with microcephaly in six Korean patients: When to consider EFTUD2 analysis?Eur J Med Genet. 2022 May;65(5):104478. doi: 10.1016/j.ejmg.2022.104478. Epub 2022 Apr 5. Eur J Med Genet. 2022. PMID: 35395430
-
[Clinical case analysis and literature review of mandibulofacial dysostosis with microcephaly syndrome].Lin Chuang Er Bi Yan Hou Tou Jing Wai Ke Za Zhi. 2022 Jan;36(1):36-40. doi: 10.13201/j.issn.2096-7993.2022.01.008. Lin Chuang Er Bi Yan Hou Tou Jing Wai Ke Za Zhi. 2022. PMID: 34979617 Free PMC article. Review. Chinese.
Cited by
-
"Mandibulofacial dysostosis with microcephaly" caused by EFTUD2 mutations: expanding the phenotype.Am J Med Genet A. 2013 Jan;161A(1):108-13. doi: 10.1002/ajmg.a.35696. Epub 2012 Dec 14. Am J Med Genet A. 2013. PMID: 23239648 Free PMC article.
-
A common cellular response to broad splicing perturbations is characterized by metabolic transcript downregulation driven by the Mdm2-p53 axis.Dis Model Mech. 2024 Feb 1;17(2):dmm050356. doi: 10.1242/dmm.050356. Epub 2024 Mar 1. Dis Model Mech. 2024. PMID: 38426258 Free PMC article.
-
Spliceosomopathies: Diseases and mechanisms.Dev Dyn. 2020 Sep;249(9):1038-1046. doi: 10.1002/dvdy.214. Epub 2020 Jun 29. Dev Dyn. 2020. PMID: 32506634 Free PMC article. Review.
-
Facial dysostoses: Etiology, pathogenesis and management.Am J Med Genet C Semin Med Genet. 2013 Nov;163C(4):283-94. doi: 10.1002/ajmg.c.31375. Epub 2013 Oct 4. Am J Med Genet C Semin Med Genet. 2013. PMID: 24123981 Free PMC article. Review.
-
FORGE Canada Consortium: outcomes of a 2-year national rare-disease gene-discovery project.Am J Hum Genet. 2014 Jun 5;94(6):809-17. doi: 10.1016/j.ajhg.2014.05.003. Am J Hum Genet. 2014. PMID: 24906018 Free PMC article.
References
-
- The Treacher Collins Syndrome Collaborative Group Positional cloning of a gene involved in the pathogenesis of Treacher Collins syndrome. Nat. Genet. 1996;12:130–136. - PubMed
-
- Dauwerse J.G., Dixon J., Seland S., Ruivenkamp C.A., van Haeringen A., Hoefsloot L.H., Peters D.J., Boers A.C., Daumer-Haas C., Maiwald R. Mutations in genes encoding subunits of RNA polymerases I and III cause Treacher Collins syndrome. Nat. Genet. 2011;43:20–22. - PubMed
-
- Guion-Almeida M.L., Zechi-Ceide R.M., Vendramini S., Tabith Júnior A. A new syndrome with growth and mental retardation, mandibulofacial dysostosis, microcephaly, and cleft palate. Clin. Dysmorphol. 2006;15:171–174. - PubMed
-
- Wieczorek D., Gener B., González M.J., Seland S., Fischer S., Hehr U., Kuechler A., Hoefsloot L.H., de Leeuw N., Gillessen-Kaesbach G., Lohmann D.R. Microcephaly, microtia, preauricular tags, choanal atresia and developmental delay in three unrelated patients: a mandibulofacial dysostosis distinct from Treacher Collins syndrome. Am. J. Med. Genet. A. 2009;149A:837–843. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
