

Whole-exome-sequencing identifies mutations in histone acetyltransferase gene KAT6B in individuals with the Say-Barber-Biesecker variant of Ohdo syndrome
- PMID: 22077973
- PMCID: PMC3213399
- DOI: 10.1016/j.ajhg.2011.10.008
Whole-exome-sequencing identifies mutations in histone acetyltransferase gene KAT6B in individuals with the Say-Barber-Biesecker variant of Ohdo syndrome
Abstract
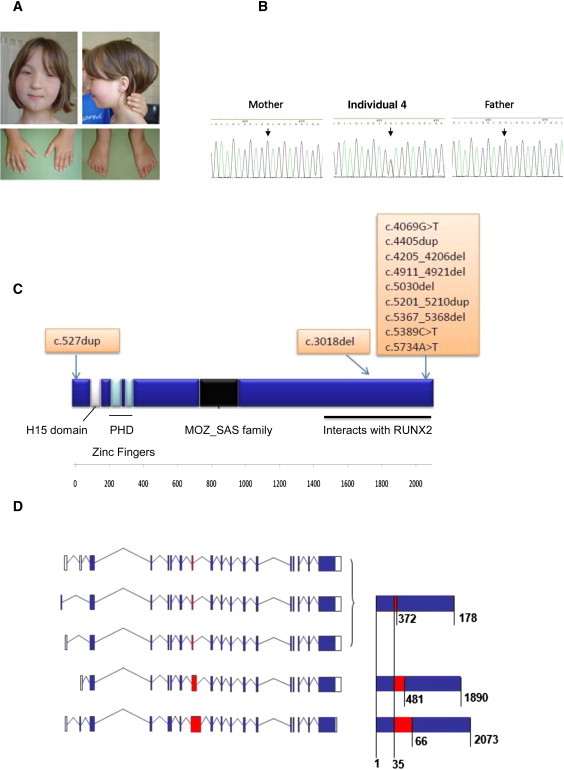
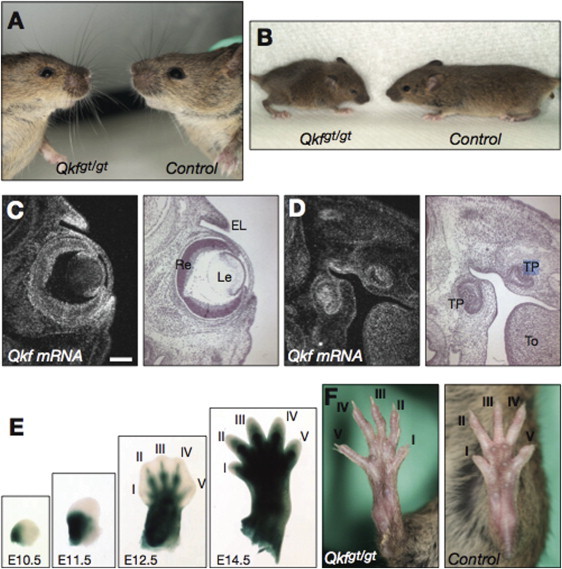
Say-Barber-Biesecker-Young-Simpson syndrome (SBBYSS or Ohdo syndrome) is a multiple anomaly syndrome characterized by severe intellectual disability, blepharophimosis, and a mask-like facial appearance. A number of individuals with SBBYSS also have thyroid abnormalities and cleft palate. The condition usually occurs sporadically and is therefore presumed to be due in most cases to new dominant mutations. In individuals with SBBYSS, a whole-exome sequencing approach was used to demonstrate de novo protein-truncating mutations in the highly conserved histone acetyltransferase gene KAT6B (MYST4/MORF)) in three out of four individuals sequenced. Sanger sequencing was used to confirm truncating mutations of KAT6B, clustering in the final exon of the gene in all four individuals and in a further nine persons with typical SBBYSS. Where parental samples were available, the mutations were shown to have occurred de novo. During mammalian development KAT6B is upregulated specifically in the developing central nervous system, facial structures, and limb buds. The phenotypic features seen in the Qkf mouse, a hypomorphic Kat6b mutant, include small eyes, ventrally placed ears and long first digits that mirror the human phenotype. This is a further example of how perturbation of a protein involved in chromatin modification might give rise to a multisystem developmental disorder.
Copyright © 2011 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
A novel truncating variant within exon 7 of KAT6B associated with features of both Say-Barber-Bieseker-Young-Simpson syndrome and genitopatellar syndrome: Further evidence of a continuum in the clinical spectrum of KAT6B-related disorders.Am J Med Genet A. 2018 Feb;176(2):455-459. doi: 10.1002/ajmg.a.38571. Epub 2017 Dec 11. Am J Med Genet A. 2018. PMID: 29226580
-
De Novo Mutation of KAT6B Gene Causing Atypical Say-Barber-Biesecker-Young-Simpson Syndrome or Genitopatellar Syndrome.Fetal Pediatr Pathol. 2017 Apr;36(2):130-138. doi: 10.1080/15513815.2017.1281364. Epub 2017 Feb 7. Fetal Pediatr Pathol. 2017. PMID: 28426343
-
De novo mutations of the gene encoding the histone acetyltransferase KAT6B in two patients with Say-Barber/Biesecker/Young-Simpson syndrome.Am J Med Genet A. 2013 Apr;161A(4):884-8. doi: 10.1002/ajmg.a.35848. Epub 2013 Feb 22. Am J Med Genet A. 2013. PMID: 23436491
-
The KAT6B-related disorders genitopatellar syndrome and Ohdo/SBBYS syndrome have distinct clinical features reflecting distinct molecular mechanisms.Hum Mutat. 2012 Nov;33(11):1520-5. doi: 10.1002/humu.22141. Epub 2012 Jul 12. Hum Mutat. 2012. PMID: 22715153 Free PMC article. Review.
-
Say-Barber-Biesecker-Young-Simpson syndrome and Genitopatellar syndrome: Lumping or splitting?Clin Genet. 2019 Feb;95(2):253-261. doi: 10.1111/cge.13127. Epub 2018 Jan 25. Clin Genet. 2019. PMID: 28857140 Review.
Cited by
-
De novo mutations in MLL cause Wiedemann-Steiner syndrome.Am J Hum Genet. 2012 Aug 10;91(2):358-64. doi: 10.1016/j.ajhg.2012.06.008. Epub 2012 Jul 12. Am J Hum Genet. 2012. PMID: 22795537 Free PMC article.
-
Further delineation of the clinical spectrum of KAT6B disorders and allelic series of pathogenic variants.Genet Med. 2020 Aug;22(8):1338-1347. doi: 10.1038/s41436-020-0811-8. Epub 2020 May 19. Genet Med. 2020. PMID: 32424177 Free PMC article. Review.
-
Expanding the clinical and mutational spectrum of Kaufman oculocerebrofacial syndrome with biallelic UBE3B mutations.Hum Genet. 2014 Jul;133(7):939-49. doi: 10.1007/s00439-014-1436-2. Epub 2014 Mar 11. Hum Genet. 2014. PMID: 24615390
-
Genitopatellar syndrome: the first reported case in Japan.Hum Genome Var. 2018 May 28;5:8. doi: 10.1038/s41439-018-0010-1. eCollection 2018. Hum Genome Var. 2018. PMID: 29899993 Free PMC article.
-
The Chromatin Regulator BRPF3 Preferentially Activates the HBO1 Acetyltransferase but Is Dispensable for Mouse Development and Survival.J Biol Chem. 2016 Feb 5;291(6):2647-63. doi: 10.1074/jbc.M115.703041. Epub 2015 Dec 16. J Biol Chem. 2016. PMID: 26677226 Free PMC article.
References
-
- Verloes A., Bremond-Gignac C., Isidor B., David A., Baumann C., Leroy M.A., Stevens R., Gillerot Y., Héron D., Héron B., et al. Blepharophimosis-mental retardation syndromes: A proposed clinical classification of the so-called Ohdo syndrome, and delineation of two new BMR syndromes, one X-linked and one autosomal recessive. Am. J. Med. Genet. A. 2006;140:1285–1296. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
