

Novel SCN1A mutation in a proband with malignant migrating partial seizures of infancy
- PMID: 21555645
- PMCID: PMC3092158
- DOI: 10.1001/archneurol.2011.98
Novel SCN1A mutation in a proband with malignant migrating partial seizures of infancy
Abstract
Objective: To characterize a novel SCN1A mutation in a proband with malignant migrating partial seizures of infancy.
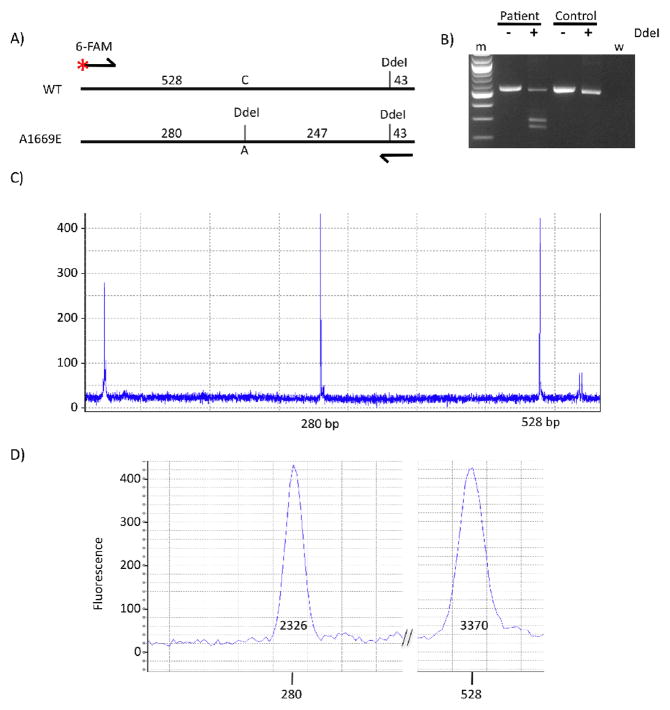
Design: Genomic DNA was isolated from blood and submitted for commercial testing. The identified missense mutation was confirmed in brain DNA obtained at autopsy. Genomic DNA from the brain of the proband was analyzed by comparative genome hybridization, and the coding exons of SCN9A were amplified. Quantitation studies of the mutant transcript were performed.
Setting: Children's National Medical Center and Yale University School of Medicine. PROBAND: A full-term female infant who experienced seizure onset at age 10 weeks, with progression of hemiclonic, apneic, and multifocal migrating partial seizures leading to recurrent status epilepticus and death at age 9 months.
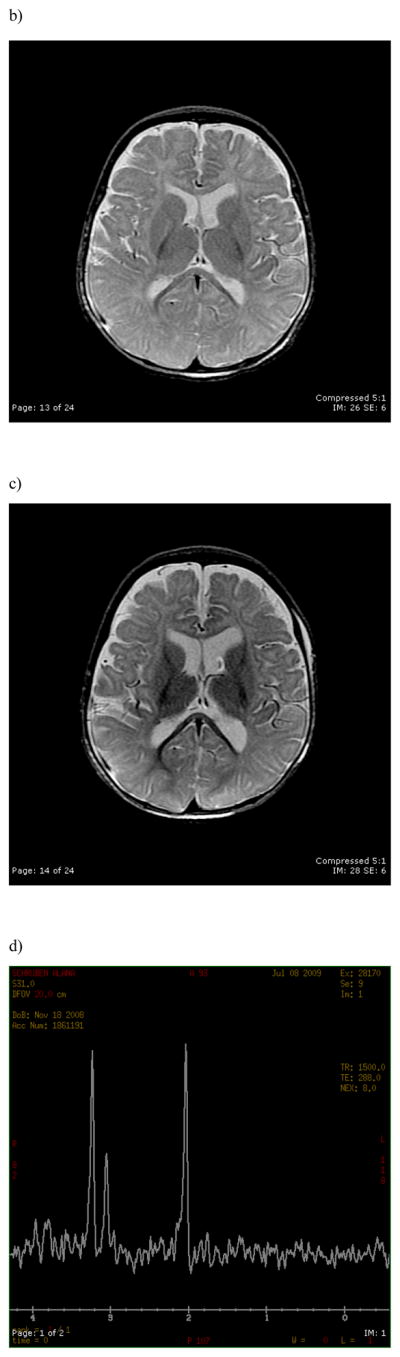
Main outcome measures: Electroencephalographic and magnetic resonance imaging results, quantitative RNA expression, and secondary mutation test results.
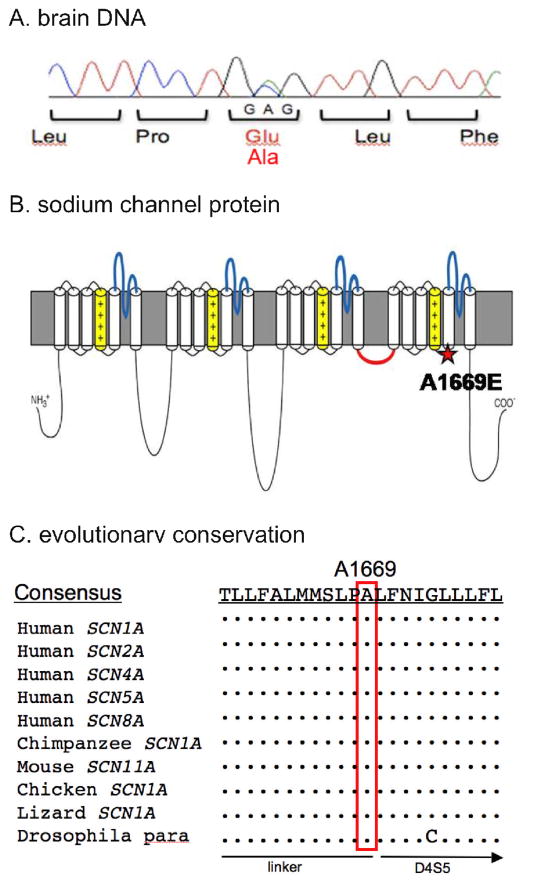
Results: The heterozygous missense mutation c.C5006C>A was identified by sequencing genomic DNA from blood and was confirmed in brain DNA. The resulting amino acid substitution p.A1669E alters an evolutionarily conserved residue in an intracellular linker of domain 4 of the SCN1A sodium channel protein Na(v)1.1. The mutant transcript is found to be expressed at levels comparable to the wild-type allele in brain RNA. No variation in copy number was detected in the chromosome region 2q24 containing SCN1A or elsewhere in the genome. No mutations were detected in the linked sodium channel gene SCN9A, which has been reported to act as a modifier of SCN1A mutations.
Conclusion: This report expands the spectrum of SCN1A epileptic channelopathies to include malignant migrating partial seizures of infancy.
Figures
Similar articles
-
Epilepsy phenotype associated with a chromosome 2q24.3 deletion involving SCN1A: Migrating partial seizures of infancy or atypical Dravet syndrome?Epilepsy Res. 2015 Jan;109:34-9. doi: 10.1016/j.eplepsyres.2014.10.008. Epub 2014 Oct 28. Epilepsy Res. 2015. PMID: 25524840
-
De novo SCN1A mutations in migrating partial seizures of infancy.Neurology. 2011 Jul 26;77(4):380-3. doi: 10.1212/WNL.0b013e318227046d. Epub 2011 Jul 13. Neurology. 2011. PMID: 21753172 Free PMC article.
-
A role of SCN9A in human epilepsies, as a cause of febrile seizures and as a potential modifier of Dravet syndrome.PLoS Genet. 2009 Sep;5(9):e1000649. doi: 10.1371/journal.pgen.1000649. Epub 2009 Sep 18. PLoS Genet. 2009. PMID: 19763161 Free PMC article.
-
Effect of localization of missense mutations in SCN1A on epilepsy phenotype severity.Neurology. 2004 Jul 27;63(2):329-34. doi: 10.1212/01.wnl.0000129829.31179.5b. Neurology. 2004. PMID: 15277629 Review.
-
Elicited repetitive daily blindness: a new phenotype associated with hemiplegic migraine and SCN1A mutations.Neurology. 2009 Mar 31;72(13):1178-83. doi: 10.1212/01.wnl.0000345393.53132.8c. Neurology. 2009. PMID: 19332696 Review.
Cited by
-
Migrating partial seizures of infancy: expansion of the electroclinical, radiological and pathological disease spectrum.Brain. 2013 May;136(Pt 5):1578-91. doi: 10.1093/brain/awt073. Epub 2013 Apr 18. Brain. 2013. PMID: 23599387 Free PMC article.
-
Rapid evolution of a voltage-gated sodium channel gene in a lineage of electric fish leads to a persistent sodium current.PLoS Biol. 2018 Mar 27;16(3):e2004892. doi: 10.1371/journal.pbio.2004892. eCollection 2018 Mar. PLoS Biol. 2018. PMID: 29584718 Free PMC article.
-
SLC25A22 is a novel gene for migrating partial seizures in infancy.Ann Neurol. 2013 Dec;74(6):873-82. doi: 10.1002/ana.23998. Ann Neurol. 2013. PMID: 24596948 Free PMC article.
-
Electroencephalogram of age-dependent epileptic encephalopathies in infancy and early childhood.Epilepsy Res Treat. 2013;2013:743203. doi: 10.1155/2013/743203. Epub 2013 Aug 19. Epilepsy Res Treat. 2013. PMID: 24024028 Free PMC article. Review.
-
SCN1A Mutation-Beyond Dravet Syndrome: A Systematic Review and Narrative Synthesis.Front Neurol. 2021 Dec 24;12:743726. doi: 10.3389/fneur.2021.743726. eCollection 2021. Front Neurol. 2021. PMID: 35002916 Free PMC article. Review.
References
-
- Coppola G, Plouin P, Chiron C, Robain O, Dulac O. Migrating partial seizures in infancy: A malignant disorder with developmental arrest. Epilepsia. 1995;36:1017–1024. - PubMed
-
- Coppola G. Malignant migrating partial seizures in infancy: An epilepsy syndrome of unknown etiology. Epilepsia. 2009;50:49–51. - PubMed
-
- Marsh E, Melamed S, Barron T, Clancy R. Migrating partial seizures in infancy: Expanding the phenotype of a rare seizure syndrome. Epilepsia. 2005;46:568–572. - PubMed
-
- Gross-Tsur V, Ben-Zeev B, Shalev RS. Malignant migrating partial seizures in infancy. Pediatr Neurol. 2004;31:287–290. - PubMed
-
- Wilmhurst JM, Appleton DB, Grattan-Smith PJ. Migrating Partial Seizures in Infancy: Two New Cases. J Child Neurol. 2000;15:717–722. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
