

Mutations in C16orf57 and normal-length telomeres unify a subset of patients with dyskeratosis congenita, poikiloderma with neutropenia and Rothmund-Thomson syndrome
- PMID: 20817924
- PMCID: PMC2957322
- DOI: 10.1093/hmg/ddq371
Mutations in C16orf57 and normal-length telomeres unify a subset of patients with dyskeratosis congenita, poikiloderma with neutropenia and Rothmund-Thomson syndrome
Abstract
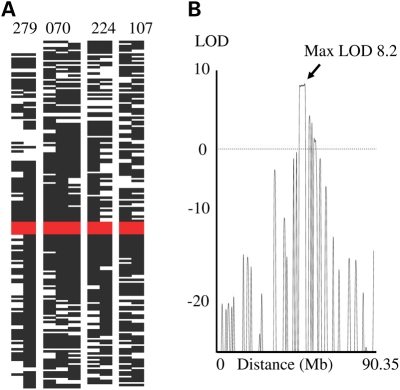
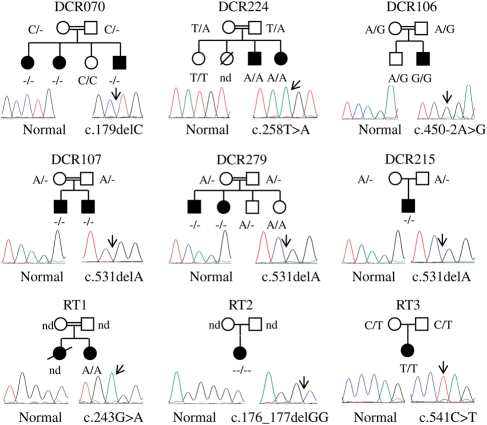
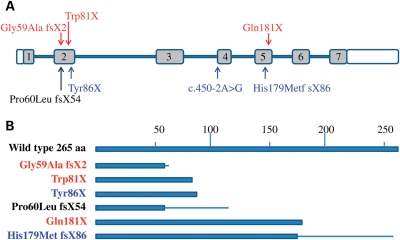
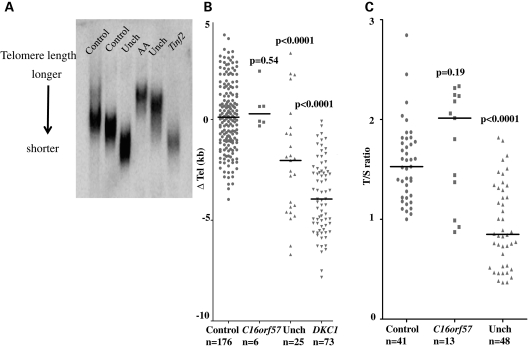
Dyskeratosis congenita (DC) is an inherited poikiloderma which in addition to the skin abnormalities is typically associated with nail dystrophy, leucoplakia, bone marrow failure, cancer predisposition and other features. Approximately 50% of DC patients remain genetically uncharacterized. All the DC genes identified to date are important in telomere maintenance. To determine the genetic basis of the remaining cases of DC, we undertook linkage analysis in 20 families and identified a common candidate gene region on chromosome 16 in a subset of these. This region included the C16orf57 gene recently identified to be mutated in poikiloderma with neutropenia (PN), an inherited poikiloderma displaying significant clinical overlap with DC. Analysis of the C16orf57 gene in our uncharacterized DC patients revealed homozygous mutations in 6 of 132 families. In addition, three of six families previously classified as Rothmund-Thomson syndrome (RTS-a poikiloderma that is sometimes confused with PN) were also found to have homozygous C16orf57 mutations. Given the role of the previous DC genes in telomere maintenance, telomere length was analysed in these patients and found to be comparable to age-matched controls. These findings suggest that mutations in C16orf57 unify a distinct set of families which clinically can be categorized as DC, PN or RTS. This study also highlights the multi-system nature (wider than just poikiloderma and neutropenia) of the clinical features of affected individuals (and therefore house-keeping function of C16orf57), a possible role for C16orf57 in apoptosis, as well as a distinct difference from previously characterized DC patients because telomere length was normal.
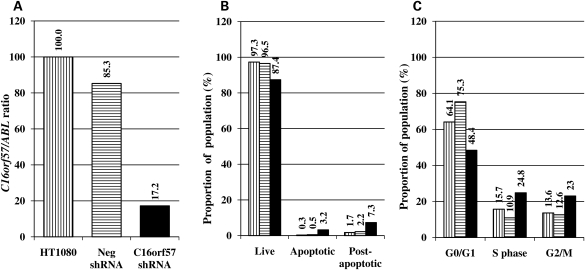
Figures
Similar articles
-
[Mutations of C16orf57 gene have been identified in the poikiloderma-neutropenia syndrome and in a specific subset of congenital dyskeratosis with normal-length telomeres].Ann Dermatol Venereol. 2011;138(4):362-3. doi: 10.1016/j.annder.2011.01.026. Epub 2011 Feb 26. Ann Dermatol Venereol. 2011. PMID: 21497268 French. No abstract available.
-
C16orf57, a gene mutated in poikiloderma with neutropenia, encodes a putative phosphodiesterase responsible for the U6 snRNA 3' end modification.Genes Dev. 2012 Sep 1;26(17):1911-25. doi: 10.1101/gad.193169.112. Epub 2012 Aug 16. Genes Dev. 2012. PMID: 22899009 Free PMC article.
-
Systematic search for neutropenia should be part of the first screening in patients with poikiloderma.Eur J Med Genet. 2012 Jan;55(1):8-11. doi: 10.1016/j.ejmg.2011.07.004. Epub 2011 Aug 18. Eur J Med Genet. 2012. PMID: 21872685
-
Recent progress in dyskeratosis congenita.Int J Hematol. 2010 Oct;92(3):419-24. doi: 10.1007/s12185-010-0695-5. Epub 2010 Oct 1. Int J Hematol. 2010. PMID: 20882440 Review.
-
Rothmund-Thomson syndrome.Orphanet J Rare Dis. 2010 Jan 29;5:2. doi: 10.1186/1750-1172-5-2. Orphanet J Rare Dis. 2010. PMID: 20113479 Free PMC article. Review.
Cited by
-
Incomplete splicing of neutrophil-specific genes affects neutrophil development in a zebrafish model of poikiloderma with neutropenia.RNA Biol. 2015;12(4):426-34. doi: 10.1080/15476286.2015.1017240. RNA Biol. 2015. PMID: 25849198 Free PMC article.
-
Mutations in the telomere capping complex in bone marrow failure and related syndromes.Haematologica. 2013 Mar;98(3):334-8. doi: 10.3324/haematol.2012.071068. Epub 2012 Aug 16. Haematologica. 2013. PMID: 22899577 Free PMC article.
-
Identification of a novel C16orf57 mutation in Athabaskan patients with Poikiloderma with Neutropenia.Am J Med Genet A. 2011 Feb;155A(2):337-42. doi: 10.1002/ajmg.a.33807. Epub 2010 Dec 22. Am J Med Genet A. 2011. PMID: 21271650 Free PMC article.
-
Differences in disease severity but similar telomere lengths in genetic subgroups of patients with telomerase and shelterin mutations.PLoS One. 2011;6(9):e24383. doi: 10.1371/journal.pone.0024383. Epub 2011 Sep 13. PLoS One. 2011. PMID: 21931702 Free PMC article.
-
Constitutional mutations in RTEL1 cause severe dyskeratosis congenita.Am J Hum Genet. 2013 Mar 7;92(3):448-53. doi: 10.1016/j.ajhg.2013.02.001. Epub 2013 Feb 28. Am J Hum Genet. 2013. PMID: 23453664 Free PMC article.
References
-
- Dokal I. Dyskeratosis congenita in all its forms. Br. J. Haematol. 2000;110:768–779. doi:10.1046/j.1365-2141.2000.02109.x. - DOI - PubMed
-
- Heiss N.S., Knight S.W., Vulliamy T.J., Klauck S.M., Wiemann S., Mason P.J., Poustka A., Dokal I. X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat. Genet. 1998;19:32–38. doi:10.1038/ng0598-32. - DOI - PubMed
-
- Vulliamy T., Marrone A., Goldman F., Dearlove A., Bessler M., Mason P.J., Dokal I. The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature. 2001;413:432–435. doi:10.1038/35096585. - DOI - PubMed
-
- Armanios M., Chen J.L., Chang Y.P., Brodsky R.A., Hawkins A., Griffin C.A., Eshleman J.R., Cohen A.R., Chakravarti A., Hamosh A., et al. Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita. Proc. Natl Acad. Sci. USA. 2005;102:15960–15964. doi:10.1073/pnas.0508124102. - DOI - PMC - PubMed
-
- Walne A.J., Vulliamy T., Marrone A., Beswick R., Kirwan M., Masunari Y., Al-Qurashi F.H., Aljurf M., Dokal I. Genetic heterogeneity in autosomal recessive dyskeratosis congenita with one subtype due to mutations in the telomerase-associated protein NOP10. Hum. Mol. Genet. 2007;16:1619–1629. doi:10.1093/hmg/ddm111. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
