

TUBA1A mutations cause wide spectrum lissencephaly (smooth brain) and suggest that multiple neuronal migration pathways converge on alpha tubulins
- PMID: 20466733
- PMCID: PMC2893812
- DOI: 10.1093/hmg/ddq182
TUBA1A mutations cause wide spectrum lissencephaly (smooth brain) and suggest that multiple neuronal migration pathways converge on alpha tubulins
Abstract
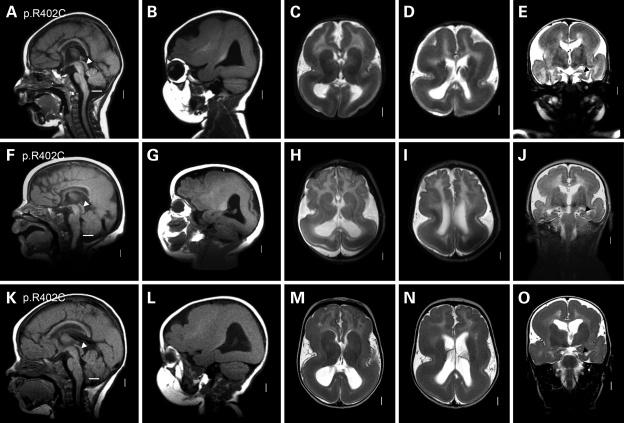
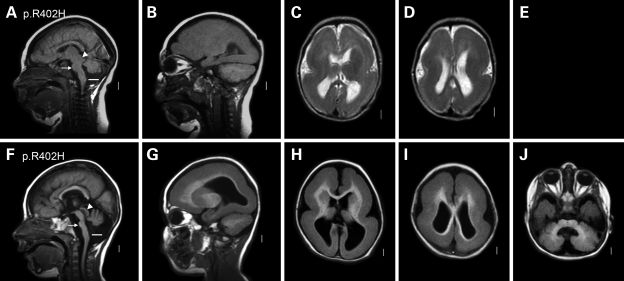
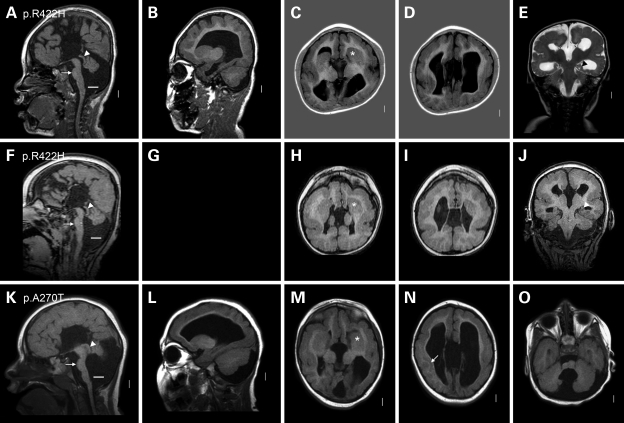
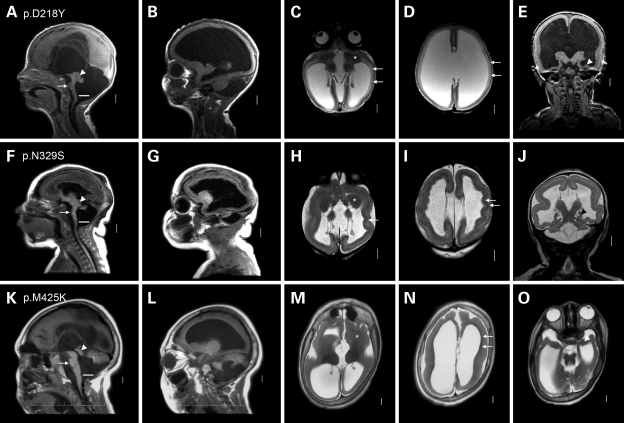
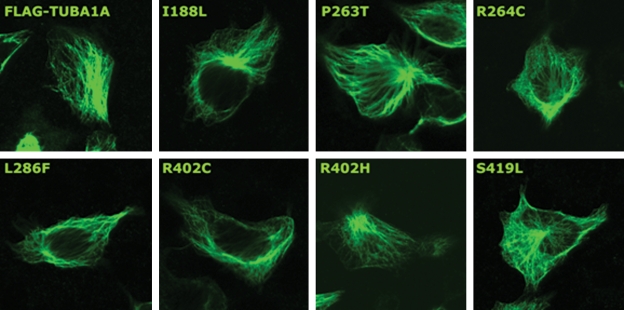
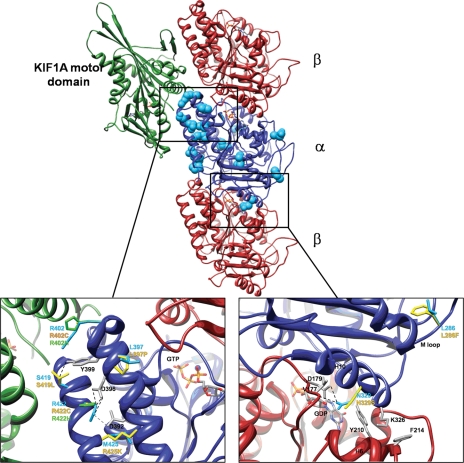
We previously showed that mutations in LIS1 and DCX account for approximately 85% of patients with the classic form of lissencephaly (LIS). Some rare forms of LIS are associated with a disproportionately small cerebellum, referred to as lissencephaly with cerebellar hypoplasia (LCH). Tubulin alpha1A (TUBA1A), encoding a critical structural subunit of microtubules, has recently been implicated in LIS. Here, we screen the largest cohort of unexplained LIS patients examined to date to determine: (i) the frequency of TUBA1A mutations in patients with lissencephaly, (ii) the spectrum of phenotypes associated with TUBA1A mutations and (iii) the functional consequences of different TUBA1A mutations on microtubule function. We identified novel and recurrent TUBA1A mutations in approximately 1% of children with classic LIS and in approximately 30% of children with LCH, making this the first major gene associated with the rare LCH phenotype. We also unexpectedly found a TUBA1A mutation in one child with agenesis of the corpus callosum and cerebellar hypoplasia without LIS. Thus, our data demonstrate a wider spectrum of phenotypes than previously reported and allow us to propose new recommendations for clinical testing. We also provide cellular and structural data suggesting that LIS-associated mutations of TUBA1A operate via diverse mechanisms that include disruption of binding sites for microtubule-associated proteins (MAPs).
Figures
Similar articles
-
Refining the phenotype of alpha-1a Tubulin (TUBA1A) mutation in patients with classical lissencephaly.Clin Genet. 2008 Nov;74(5):425-33. doi: 10.1111/j.1399-0004.2008.01093.x. Clin Genet. 2008. PMID: 18954413
-
TUBA1A tubulinopathy mutants disrupt neuron morphogenesis and override XMAP215/Stu2 regulation of microtubule dynamics.Elife. 2022 May 5;11:e76189. doi: 10.7554/eLife.76189. Elife. 2022. PMID: 35511030 Free PMC article.
-
Neuropathological phenotype of a distinct form of lissencephaly associated with mutations in TUBA1A.Brain. 2008 Sep;131(Pt 9):2304-20. doi: 10.1093/brain/awn155. Epub 2008 Jul 18. Brain. 2008. PMID: 18669490
-
TUBA1A mutation-associated lissencephaly: case report and review of the literature.Pediatr Neurol. 2012 Feb;46(2):127-31. doi: 10.1016/j.pediatrneurol.2011.11.017. Pediatr Neurol. 2012. PMID: 22264709 Review.
-
Tubulin mutations in human neurodevelopmental disorders.Semin Cell Dev Biol. 2023 Mar 15;137:87-95. doi: 10.1016/j.semcdb.2022.07.009. Epub 2022 Jul 30. Semin Cell Dev Biol. 2023. PMID: 35915025 Review.
Cited by
-
Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with a loss-of-function mutation in CDK5.Hum Genet. 2015 Mar;134(3):305-14. doi: 10.1007/s00439-014-1522-5. Epub 2015 Jan 6. Hum Genet. 2015. PMID: 25560765
-
Identification of a 31-bp deletion in the RELN gene causing lissencephaly with cerebellar hypoplasia in sheep.PLoS One. 2013 Nov 19;8(11):e81072. doi: 10.1371/journal.pone.0081072. eCollection 2013. PLoS One. 2013. PMID: 24260534 Free PMC article.
-
Reduced TUBA1A Tubulin Causes Defects in Trafficking and Impaired Adult Motor Behavior.eNeuro. 2020 Apr 27;7(2):ENEURO.0045-20.2020. doi: 10.1523/ENEURO.0045-20.2020. Print 2020 Mar/Apr. eNeuro. 2020. PMID: 32184299 Free PMC article.
-
Microtubule dynamics in axon guidance.Neurosci Bull. 2014 Aug;30(4):569-83. doi: 10.1007/s12264-014-1444-6. Epub 2014 Jun 26. Neurosci Bull. 2014. PMID: 24968808 Free PMC article. Review.
-
Mutation of the α-tubulin Tuba1a leads to straighter microtubules and perturbs neuronal migration.J Cell Biol. 2017 Aug 7;216(8):2443-2461. doi: 10.1083/jcb.201607074. Epub 2017 Jul 7. J Cell Biol. 2017. PMID: 28687665 Free PMC article.
References
-
- Dobyns W.B., Truwit C.L., Ross M.E., Matsumoto N., Pilz D.T., Ledbetter D.H., Gleeson J.G., Walsh C.A., Barkovich A.J. Differences in the gyral pattern distinguish chromosome 17-linked and X-linked lissencephaly. Neurology. 1999;53:270–277. - PubMed
-
- Pilz D.T., Macha M.E., Precht K.S., Smith A.C., Dobyns W.B., Ledbetter D.H. Fluorescence in situ hybridization analysis with LIS1 specific probes reveals a high deletion mutation rate in isolated lissencephaly sequence. Genet. Med. 1998;1:29–33. - PubMed
-
- Pilz D.T., Matsumoto N., Minnerath S., Mills P., Gleeson J.G., Allen K.M., Walsh C.A., Barkovich A.J., Dobyns W.B., Ledbetter D.H., et al. LIS1 and XLIS (DCX) mutations cause most classical lissencephaly, but different patterns of malformation. Hum. Mol. Genet. 1998;7:2029–2037. doi:10.1093/hmg/7.13.2029. - DOI - PubMed
-
- Forman M.S., Squier W., Dobyns W.B., Golden J.A. Genotypically defined lissencephalies show distinct pathologies. J. Neuropathol. Exp. Neurol. 2005;64:847–857. doi:10.1097/01.jnen.0000182978.56612.41. - DOI - PubMed
-
- Ross M.E., Swanson K., Dobyns W.B. Lissencephaly with cerebellar hypoplasia (LCH): a heterogeneous group of cortical malformations. Neuropediatrics. 2001;32:256–263. doi:10.1055/s-2001-19120. - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
