

Prevalence of genetic muscle disease in Northern England: in-depth analysis of a muscle clinic population
- PMID: 19767415
- PMCID: PMC4038491
- DOI: 10.1093/brain/awp236
Prevalence of genetic muscle disease in Northern England: in-depth analysis of a muscle clinic population
Abstract
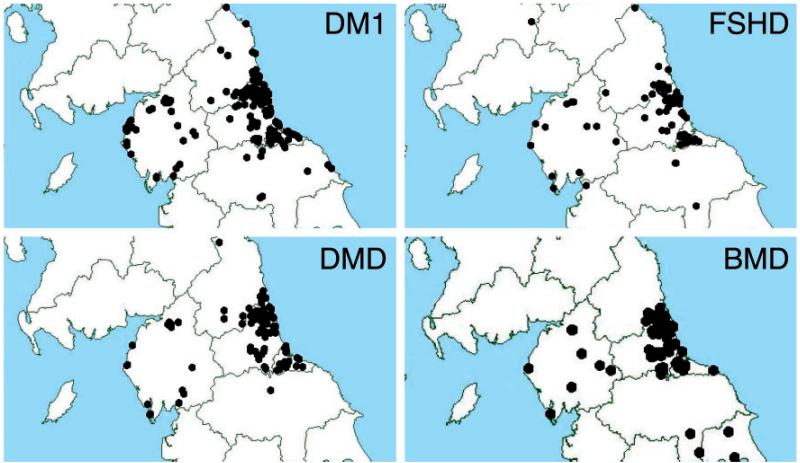
We have performed a detailed population study of patients with genetic muscle disease in the northern region of England. Our current clinic population comprises over 1100 patients in whom we have molecularly characterized 31 separate muscle disease entities. Diagnostic clarity achieved through careful delineation of clinical features supported by histological, immunological and genetic analysis has allowed us to reach a definitive diagnosis in 75.7% of our patients. We have compared our case profile with that from Walton and Nattrass' seminal study from 1954, also of the northern region, together with data from other more recent studies from around the world. Point prevalence figures for each of the five major disease categories are comparable with those from other recent studies. Myotonic dystrophies are the most common, comprising 28.6% of our clinic population with a point prevalence of 10.6/100,000. Next most frequent are the dystrophinopathies and facioscapulohumeral muscular dystrophy making up 22.9% (8.46/100,000) and 10.7% (3.95/100,000) of the clinic population, respectively. Spinal muscular atrophy patients account for 5.1% or 1.87/100,000 patients. Limb girdle muscular dystrophy, which was described for the first time in the paper by Walton and Nattrass (1954) and comprised 17% of their clinic population, comprises 6.2% of our clinic population at a combined prevalence of 2.27/100,000. The clinic population included patients with 12 other muscle disorders. These disorders ranged from a point prevalence of 0.89/100 000 for the group of congenital muscular dystrophies to conditions with only two affected individuals in a population of three million. For the first time our study provides epidemiological information for X-linked Emery-Dreifuss muscular dystrophy and the collagen VI disorders. Each of the X-linked form of Emery-Dreifuss muscular dystrophy and Ullrich muscular dystrophy has a prevalence of 0.13/100,000, making both very rare. Bethlem myopathy was relatively more common with a prevalence of 0.77/100,000. Overall our study provides comprehensive epidemiological information on individually rare inherited neuromuscular conditions in Northern England. Despite the deliberate exclusion of relatively common groups such as hereditary motor and sensory neuropathy (40/100,000) and mitochondrial disorders (9.2/100,000), the combined prevalence is 37.0/100,000, demonstrating that these disorders, taken as a group, encompass a significant proportion of patients with chronic disease. The study also illustrates the immense diagnostic progress since the first regional survey over 50 years ago by Walton and Nattrass.
Figures
Similar articles
-
Genetics and muscle pathology in the diagnosis of muscular dystrophies: An update.Indian J Pathol Microbiol. 2022 May;65(Supplement):S259-S270. doi: 10.4103/ijpm.ijpm_1074_21. Indian J Pathol Microbiol. 2022. PMID: 35562158 Review.
-
Differentiating Emery-Dreifuss muscular dystrophy and collagen VI-related myopathies using a specific CT scanner pattern.Neuromuscul Disord. 2010 Aug;20(8):517-23. doi: 10.1016/j.nmd.2010.04.009. Epub 2010 Jun 23. Neuromuscul Disord. 2010. PMID: 20576434
-
Prevalence of congenital muscular dystrophy in Italy: a population study.Neurology. 2015 Mar 3;84(9):904-11. doi: 10.1212/WNL.0000000000001303. Epub 2015 Feb 4. Neurology. 2015. PMID: 25653289 Free PMC article.
-
[Muscular dystrophies detected by immunophenotyping and genotype analysis (mRNA and DNA)].Cesk Patol. 2001 Nov;37(4):137-45. Cesk Patol. 2001. PMID: 11813630 Czech.
-
Nondystrophinopathic muscular dystrophies including myotonic dystrophy.Semin Pediatr Neurol. 1996 Jun;3(2):110-21. doi: 10.1016/s1071-9091(96)80040-4. Semin Pediatr Neurol. 1996. PMID: 8795845 Review.
Cited by
-
Rare disease clinical trials: Power in numbers.Neurol Genet. 2016 Aug 4;2(4):e92. doi: 10.1212/NXG.0000000000000092. eCollection 2016 Aug. Neurol Genet. 2016. PMID: 27540592 Free PMC article.
-
Congenital muscular dystrophies: a brief review.Semin Pediatr Neurol. 2011 Dec;18(4):277-88. doi: 10.1016/j.spen.2011.10.010. Semin Pediatr Neurol. 2011. PMID: 22172424 Free PMC article. Review.
-
Targeted Next-Generation Sequencing Reveals Mutations in Non-coding Regions and Potential Regulatory Sequences of Calpain-3 Gene in Polish Limb-Girdle Muscular Dystrophy Patients.Front Neurosci. 2021 Oct 14;15:692482. doi: 10.3389/fnins.2021.692482. eCollection 2021. Front Neurosci. 2021. PMID: 34720847 Free PMC article.
-
The collagen VI-related myopathies: muscle meets its matrix.Nat Rev Neurol. 2011 Jun 21;7(7):379-90. doi: 10.1038/nrneurol.2011.81. Nat Rev Neurol. 2011. PMID: 21691338 Free PMC article. Review.
-
Protein kinase A activation inhibits DUX4 gene expression in myotubes from patients with facioscapulohumeral muscular dystrophy.J Biol Chem. 2018 Jul 27;293(30):11837-11849. doi: 10.1074/jbc.RA118.002633. Epub 2018 Jun 13. J Biol Chem. 2018. PMID: 29899111 Free PMC article.
References
-
- Angelini C. Limb-girdle muscular dystrophies: heterogeneity of clinical phenotypes and pathogenetic mechanisms. Acta Myol. 2004;23:130–6. - PubMed
-
- Ausems MG, Verbiest J, Hermans MP, Kroos MA, Beemer FA, Wokke JH, et al. Frequency of glycogen storage disease type II in The Netherlands: implications for diagnosis and genetic counselling. Eur J Hum Genet. 1999;7:713–6. - PubMed
-
- Bachinski LL, Udd B, Meola G, Sansone V, Bassez G, Eymard B, et al. Confirmation of the type 2 myotonic dystrophy (CCTG)n expansion mutation in patients with proximal myotonic myopathy/proximal myotonic dystrophy of different European origins: a single shared haplotype indicates an ancestral founder effect. Am J Hum Genet. 2003;73:835–48. - PMC - PubMed
-
- Balci B, Aurino S, Haliloglu G, Talim B, Erdem S, Akcoren Z, et al. Calpain-3 mutations in Turkey. Eur J Pediatr. 2006;165:293–8. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
