

Mutation of DNAJC19, a human homologue of yeast inner mitochondrial membrane co-chaperones, causes DCMA syndrome, a novel autosomal recessive Barth syndrome-like condition
- PMID: 16055927
- PMCID: PMC2564511
- DOI: 10.1136/jmg.2005.036657
Mutation of DNAJC19, a human homologue of yeast inner mitochondrial membrane co-chaperones, causes DCMA syndrome, a novel autosomal recessive Barth syndrome-like condition
Abstract
Background: A novel autosomal recessive condition, dilated cardiomyopathy with ataxia (DCMA) syndrome, has been identified in the Canadian Dariusleut Hutterite population, characterised by early onset dilated cardiomyopathy with conduction defects, non-progressive cerebellar ataxia, testicular dysgenesis, growth failure, and 3-methylglutaconic aciduria.
Objective: To map DCMA syndrome and identify the mutation underlying this condition.
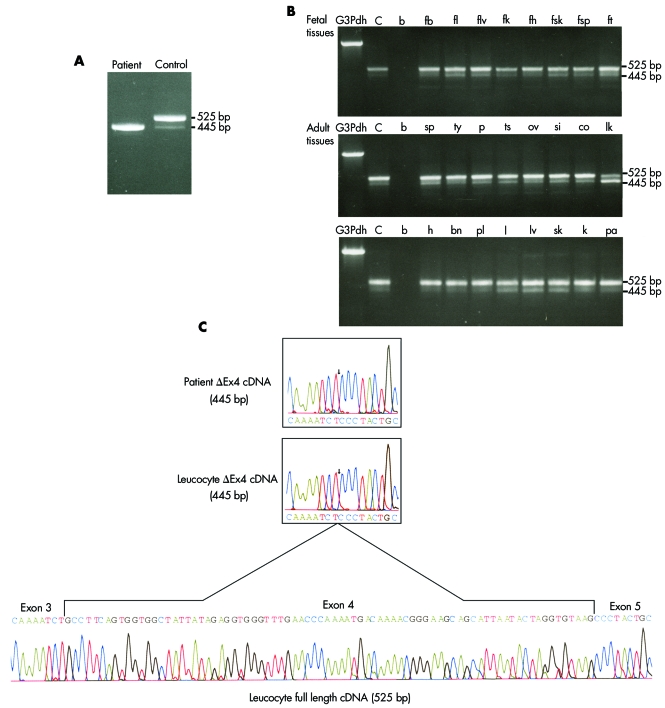
Methods: A genome wide scan was undertaken on consanguineous Hutterite families using a homozygosity mapping approach in order to identify the DCMA associated chromosomal region. Mutation analysis was carried out on positional candidate genes in this region by sequencing. Reverse transcriptase polymerase chain reaction and bioinformatics analyses were then used to characterise the mutation and determine its effect on the protein product.
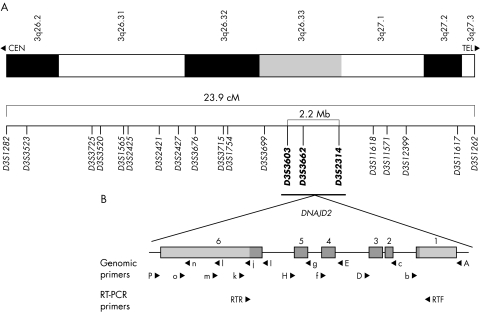
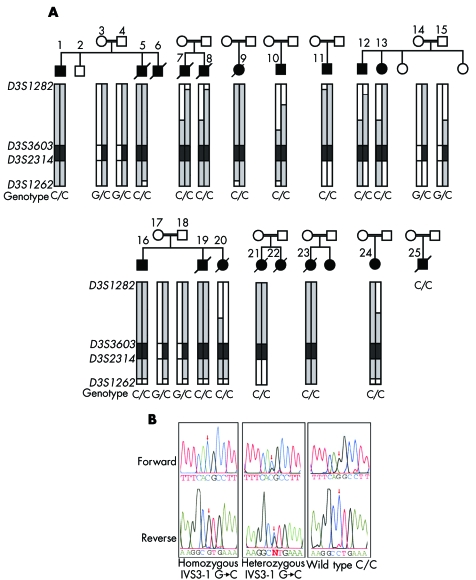
Results: The association of DCMA syndrome with a 2.2 Mb region of chromosome 3q26.33 was found. A disease associated mutation was identified: IVS3-1 G-->C in the DNAJC19 gene, encoding a DNAJ domain containing protein of previously unknown function (Entrez Gene ID 131118).
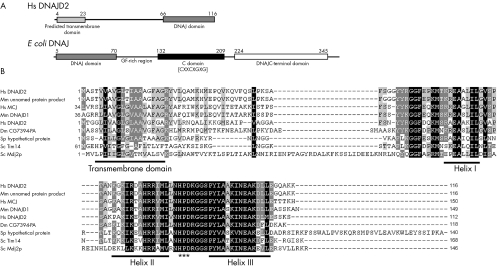
Conclusions: The DNAJC19 protein was previously localised to the mitochondria in cardiac myocytes, and shares sequence and organisational similarity with proteins from several species including two yeast mitochondrial inner membrane proteins, Mdj2p and Tim14. Tim14 is a component of the yeast inner mitochondrial membrane presequence translocase, suggesting that the unique phenotype of DCMA may be the result of defective mitochondrial protein import. It is only the second human disorder caused by defects in this pathway that has been identified.
Conflict of interest statement
Conflicts of interest: none declared
Similar articles
-
Inborn errors of metabolism with 3-methylglutaconic aciduria as discriminative feature: proper classification and nomenclature.J Inherit Metab Dis. 2013 Nov;36(6):923-8. doi: 10.1007/s10545-012-9580-0. Epub 2013 Jan 8. J Inherit Metab Dis. 2013. PMID: 23296368 Review.
-
New mutation of mitochondrial DNAJC19 causing dilated and noncompaction cardiomyopathy, anemia, ataxia, and male genital anomalies.Pediatr Res. 2012 Oct;72(4):432-7. doi: 10.1038/pr.2012.92. Epub 2012 Jul 13. Pediatr Res. 2012. PMID: 22797137
-
Cardiac features of a novel autosomal recessive dilated cardiomyopathic syndrome due to defective importation of mitochondrial protein.Cardiol Young. 2007 Apr;17(2):215-7. doi: 10.1017/S1047951107000042. Epub 2007 Jan 23. Cardiol Young. 2007. PMID: 17244376
-
The gene mutated in adult-onset type II citrullinaemia encodes a putative mitochondrial carrier protein.Nat Genet. 1999 Jun;22(2):159-63. doi: 10.1038/9667. Nat Genet. 1999. PMID: 10369257
-
Progressive Cerebellar Atrophy and a Novel Homozygous Pathogenic DNAJC19 Variant as a Cause of Dilated Cardiomyopathy Ataxia Syndrome.Pediatr Neurol. 2016 Sep;62:58-61. doi: 10.1016/j.pediatrneurol.2016.03.020. Epub 2016 Jun 4. Pediatr Neurol. 2016. PMID: 27426421 Review.
Cited by
-
Mechanisms of protein sorting in mitochondria.Cold Spring Harb Perspect Biol. 2012 Oct 1;4(10):a011320. doi: 10.1101/cshperspect.a011320. Cold Spring Harb Perspect Biol. 2012. PMID: 23028120 Free PMC article. Review.
-
Barcoding heat shock proteins to human diseases: looking beyond the heat shock response.Dis Model Mech. 2014 Apr;7(4):421-34. doi: 10.1242/dmm.014563. Dis Model Mech. 2014. PMID: 24719117 Free PMC article. Review.
-
Dilated Cardiomyopathy: A Genetic Journey from Past to Future.Int J Mol Sci. 2024 Oct 25;25(21):11460. doi: 10.3390/ijms252111460. Int J Mol Sci. 2024. PMID: 39519012 Free PMC article. Review.
-
Inborn errors of metabolism with 3-methylglutaconic aciduria as discriminative feature: proper classification and nomenclature.J Inherit Metab Dis. 2013 Nov;36(6):923-8. doi: 10.1007/s10545-012-9580-0. Epub 2013 Jan 8. J Inherit Metab Dis. 2013. PMID: 23296368 Review.
-
Left ventricular noncompaction: a disorder with genotypic and phenotypic heterogeneity-a narrative review.Cardiovasc Diagn Ther. 2022 Aug;12(4):495-515. doi: 10.21037/cdt-22-198. Cardiovasc Diagn Ther. 2022. PMID: 36033229 Free PMC article. Review.
References
-
- Hostetler J A. History and relevance of the Hutterite population for genetic studies. Am J Med Genet 198522453–362. - PubMed
-
- Barth P G, Valianpour F, Bowen V M, Lam J, Duran M, Vas F M, Wanders R J. X‐linked cardioskeletal myopathy and neutropenia (Barth syndrome): an update. Am J Med Genet A 2004126349–354. - PubMed
-
- Ly T B, Peters V, Gibson K M, Liesert M, Buckel W, Wilcken B, Carpenter K, Ensenauer R, Hoffmann G F, Mack M, Zschocke J. Mutations in the AUH gene cause 3‐methylglutaconic aciduria type I. Hum Mutat 200321401–407. - PubMed
-
- Gunay‐Aygun M. 3‐Methylglutaconic aciduria: a common biochemical marker in various syndromes with diverse clinical features. Mol Genet Metab 2005841–3. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous