

Atypical forms of incontinentia pigmenti in male individuals result from mutations of a cytosine tract in exon 10 of NEMO (IKK-gamma)
- PMID: 11179023
- PMCID: PMC1274488
- DOI: 10.1086/318806
Atypical forms of incontinentia pigmenti in male individuals result from mutations of a cytosine tract in exon 10 of NEMO (IKK-gamma)
Abstract
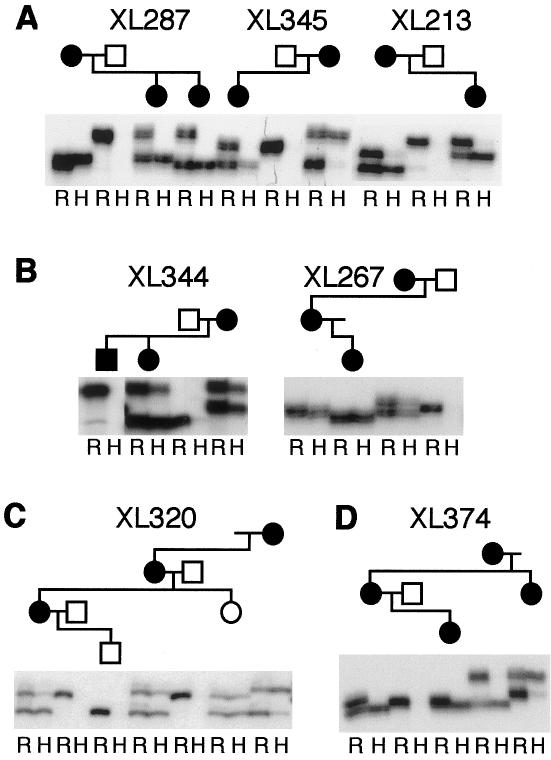
Familial incontinentia pigmenti (IP [MIM 308310]), or Bloch-Sulzberger syndrome, is an X-linked dominant and male-lethal disorder. We recently demonstrated that mutations in NEMO (IKK-gamma), which encodes a critical component of the NF-kappaB signaling pathway, were responsible for IP. Virtually all mutations eliminate the production of NEMO, causing the typical skewing of X inactivation in female individuals and lethality in male individuals, possibly through enhanced sensitivity to apoptosis. Most mutations also give rise to classic signs of IP, but, in this report, we describe two mutations in families with atypical phenotypes. Remarkably, each family included a male individual with unusual signs, including postnatal survival and either immune dysfunction or hematopoietic disturbance. We found two duplication mutations in these families, at a cytosine tract in exon 10 of NEMO, both of which remove the zinc (Zn) finger at the C-terminus of the protein. Two deletion mutations were also identified in the same tract in additional families. However, only the duplication mutations allowed male individuals to survive, and affected female individuals with duplication mutations demonstrated random or slight skewing of X inactivation. Similarly, NF-kappaB activation was diminished in the presence of duplication mutations and was completely absent in cells with deletion mutations. These results strongly indicate that male individuals can also suffer from IP caused by NEMO mutations, and we therefore urge a reevaluation of the diagnostic criteria.
Figures


Comment in
-
Female patient showing hypohidrotic ectodermal dysplasia and immunodeficiency (HED-ID).Am J Hum Genet. 2001 Sep;69(3):664-6. doi: 10.1086/323003. Am J Hum Genet. 2001. PMID: 11484156 Free PMC article. No abstract available.
Similar articles
-
A recurrent deletion in the ubiquitously expressed NEMO (IKK-gamma) gene accounts for the vast majority of incontinentia pigmenti mutations.Hum Mol Genet. 2001 Sep 15;10(19):2171-9. doi: 10.1093/hmg/10.19.2171. Hum Mol Genet. 2001. PMID: 11590134
-
Molecular analysis of the genetic defect in a large cohort of IP patients and identification of novel NEMO mutations interfering with NF-kappaB activation.Hum Mol Genet. 2004 Aug 15;13(16):1763-73. doi: 10.1093/hmg/ddh192. Epub 2004 Jun 30. Hum Mol Genet. 2004. PMID: 15229184
-
A novel X-linked disorder of immune deficiency and hypohidrotic ectodermal dysplasia is allelic to incontinentia pigmenti and due to mutations in IKK-gamma (NEMO).Am J Hum Genet. 2000 Dec;67(6):1555-62. doi: 10.1086/316914. Epub 2000 Oct 24. Am J Hum Genet. 2000. PMID: 11047757 Free PMC article.
-
Incontinentia pigmenti (Bloch-Sulzberger syndrome).Handb Clin Neurol. 2015;132:271-80. doi: 10.1016/B978-0-444-62702-5.00020-2. Handb Clin Neurol. 2015. PMID: 26564087 Review.
-
NEMO gene rearrangement (exon 4-10 deletion) and genotype-phenotype relationship in Japanese patients with incontinentia pigmenti and review of published work in Japanese patients.J Dermatol. 2013 Apr;40(4):272-6. doi: 10.1111/1346-8138.12091. Epub 2013 Feb 11. J Dermatol. 2013. PMID: 23398170 Review.
Cited by
-
Survival of male patients with incontinentia pigmenti carrying a lethal mutation can be explained by somatic mosaicism or Klinefelter syndrome.Am J Hum Genet. 2001 Dec;69(6):1210-7. doi: 10.1086/324591. Epub 2001 Oct 22. Am J Hum Genet. 2001. PMID: 11673821 Free PMC article.
-
NEMO is a key component of NF-κB- and IRF-3-dependent TLR3-mediated immunity to herpes simplex virus.J Allergy Clin Immunol. 2011 Sep;128(3):610-7.e1-4. doi: 10.1016/j.jaci.2011.04.059. Epub 2011 Jul 1. J Allergy Clin Immunol. 2011. PMID: 21722947 Free PMC article.
-
Incontinentia pigmenti: a rare genodermatosis in a male child.J Clin Diagn Res. 2015 Feb;9(2):SD06-8. doi: 10.7860/JCDR/2015/12171.5561. Epub 2015 Feb 1. J Clin Diagn Res. 2015. PMID: 25859498 Free PMC article.
-
Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target.Nat Rev Drug Discov. 2020 Aug;19(8):553-571. doi: 10.1038/s41573-020-0071-y. Epub 2020 Jul 15. Nat Rev Drug Discov. 2020. PMID: 32669658 Free PMC article.
-
Skin manifestations of inborn errors of NF-κB.Front Pediatr. 2023 Jan 17;10:1098426. doi: 10.3389/fped.2022.1098426. eCollection 2022. Front Pediatr. 2023. PMID: 36733767 Free PMC article. Review.
References
Electronic-Database Information
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for IP [MIM 308310]; for X-linked anhidrotic ectodermal dysplasia [MIM 305100]; for autosomal recessive hypohidrotic ectodermal dysplasia [MIM 224900]; for autosomal dominant hidrotic ectodermal dysplasia [MIM 129500]; for primary congenital lymphedema [MIM 153100]); for Partington syndrome II [MIM 301220]; and for Goltz syndrome [MIM 305600])
References
-
- Barkett M, Gilmore TD (1999) Control of apoptosis by Rel/NF-kappaB transcription factors. Oncogene 18:6910–6924 - PubMed
-
- Dahl MV, Matula G, Leonards R, Tuffanelli DL (1975) Incontinentia pigmenti and defective neutrophil chemotaxis. Arch Dermatol 111:1603–1605 - PubMed
-
- DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M (1997) A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature 388:548–554 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
