

Pathophysiology of human hearing loss associated with variants in myosins
- PMID: 38562617
- PMCID: PMC10982375
- DOI: 10.3389/fphys.2024.1374901
Pathophysiology of human hearing loss associated with variants in myosins
Abstract
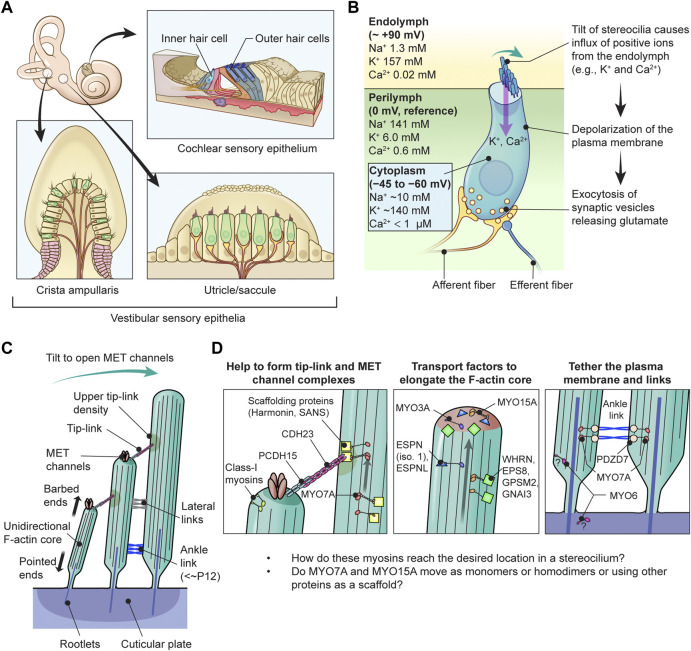
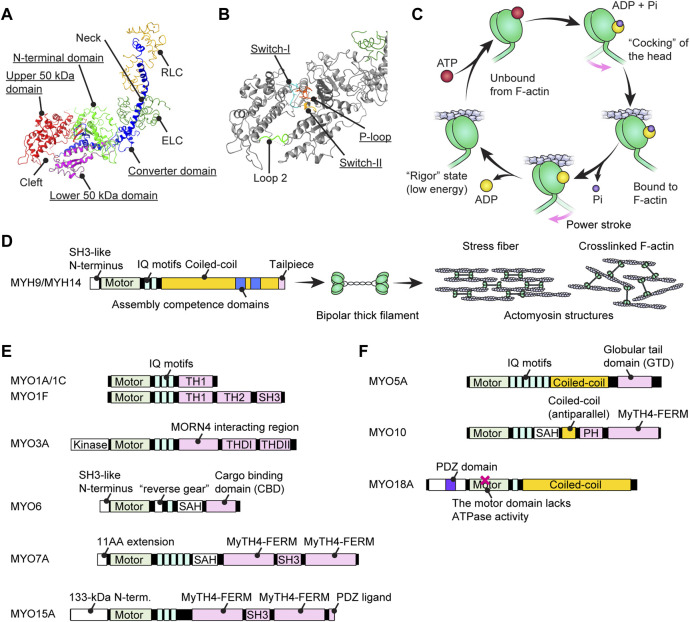
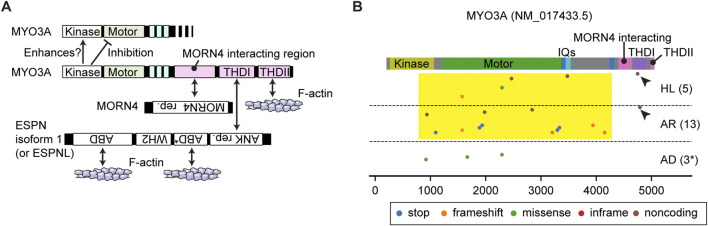
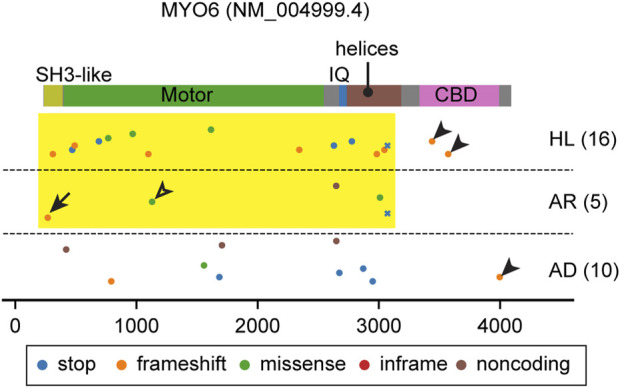
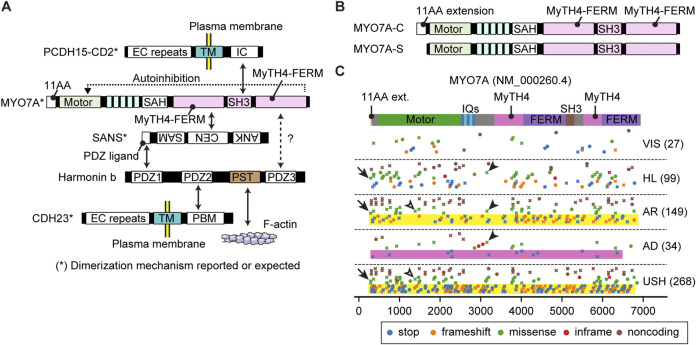
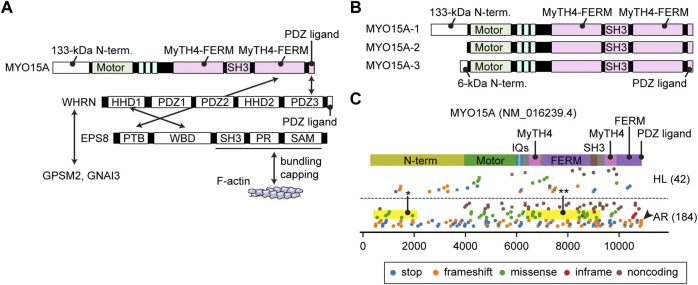
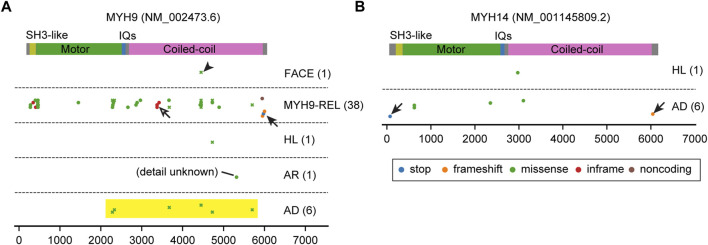
Deleterious variants of more than one hundred genes are associated with hearing loss including MYO3A, MYO6, MYO7A and MYO15A and two conventional myosins MYH9 and MYH14. Variants of MYO7A also manifest as Usher syndrome associated with dysfunction of the retina and vestibule as well as hearing loss. While the functions of MYH9 and MYH14 in the inner ear are debated, MYO3A, MYO6, MYO7A and MYO15A are expressed in inner ear hair cells along with class-I myosin MYO1C and are essential for developing and maintaining functional stereocilia on the apical surface of hair cells. Stereocilia are large, cylindrical, actin-rich protrusions functioning as biological mechanosensors to detect sound, acceleration and posture. The rigidity of stereocilia is sustained by highly crosslinked unidirectionally-oriented F-actin, which also provides a scaffold for various proteins including unconventional myosins and their cargo. Typical myosin molecules consist of an ATPase head motor domain to transmit forces to F-actin, a neck containing IQ-motifs that bind regulatory light chains and a tail region with motifs recognizing partners. Instead of long coiled-coil domains characterizing conventional myosins, the tails of unconventional myosins have various motifs to anchor or transport proteins and phospholipids along the F-actin core of a stereocilium. For these myosins, decades of studies have elucidated their biochemical properties, interacting partners in hair cells and variants associated with hearing loss. However, less is known about how myosins traffic in a stereocilium using their motor function, and how each variant correlates with a clinical condition including the severity and onset of hearing loss, mode of inheritance and presence of symptoms other than hearing loss. Here, we cover the domain structures and functions of myosins associated with hearing loss together with advances, open questions about trafficking of myosins in stereocilia and correlations between hundreds of variants in myosins annotated in ClinVar and the corresponding deafness phenotypes.
Keywords: cargo transport; hearing; hereditary deafness; myosin; stereocilia.
Copyright © 2024 Miyoshi, Belyantseva, Sajeevadathan and Friedman.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Myosins and Hearing.Adv Exp Med Biol. 2020;1239:317-330. doi: 10.1007/978-3-030-38062-5_13. Adv Exp Med Biol. 2020. PMID: 32451864 Review.
-
Functional Role of Class III Myosins in Hair Cells.Front Cell Dev Biol. 2021 Feb 25;9:643856. doi: 10.3389/fcell.2021.643856. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 33718386 Free PMC article. Review.
-
The ATPase mechanism of myosin 15, the molecular motor mutated in DFNB3 human deafness.J Biol Chem. 2021 Jan-Jun;296:100243. doi: 10.1074/jbc.RA120.014903. Epub 2021 Jan 9. J Biol Chem. 2021. PMID: 33372036 Free PMC article.
-
Unconventional myosins and the genetics of hearing loss.Am J Med Genet. 1999 Sep 24;89(3):147-57. doi: 10.1002/(sici)1096-8628(19990924)89:3<147::aid-ajmg5>3.0.co;2-6. Am J Med Genet. 1999. PMID: 10704189 Review.
-
Chaperone-enhanced purification of unconventional myosin 15, a molecular motor specialized for stereocilia protein trafficking.Proc Natl Acad Sci U S A. 2014 Aug 26;111(34):12390-5. doi: 10.1073/pnas.1409459111. Epub 2014 Aug 11. Proc Natl Acad Sci U S A. 2014. PMID: 25114250 Free PMC article.
Cited by
-
Genotype Characterization and MiRNA Expression Profiling in Usher Syndrome Cell Lines.Int J Mol Sci. 2024 Sep 17;25(18):9993. doi: 10.3390/ijms25189993. Int J Mol Sci. 2024. PMID: 39337481 Free PMC article.
-
Live-cell single-molecule fluorescence microscopy for protruding organelles reveals regulatory mechanisms of MYO7A-driven cargo transport in stereocilia of inner ear hair cells.bioRxiv [Preprint]. 2024 May 7:2024.05.04.590649. doi: 10.1101/2024.05.04.590649. bioRxiv. 2024. PMID: 38766013 Free PMC article. Preprint.
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous