

Anoctamin 5 (ANO5) Muscle Disorders: A Narrative Review
- PMID: 36292621
- PMCID: PMC9602132
- DOI: 10.3390/genes13101736
Anoctamin 5 (ANO5) Muscle Disorders: A Narrative Review
Abstract
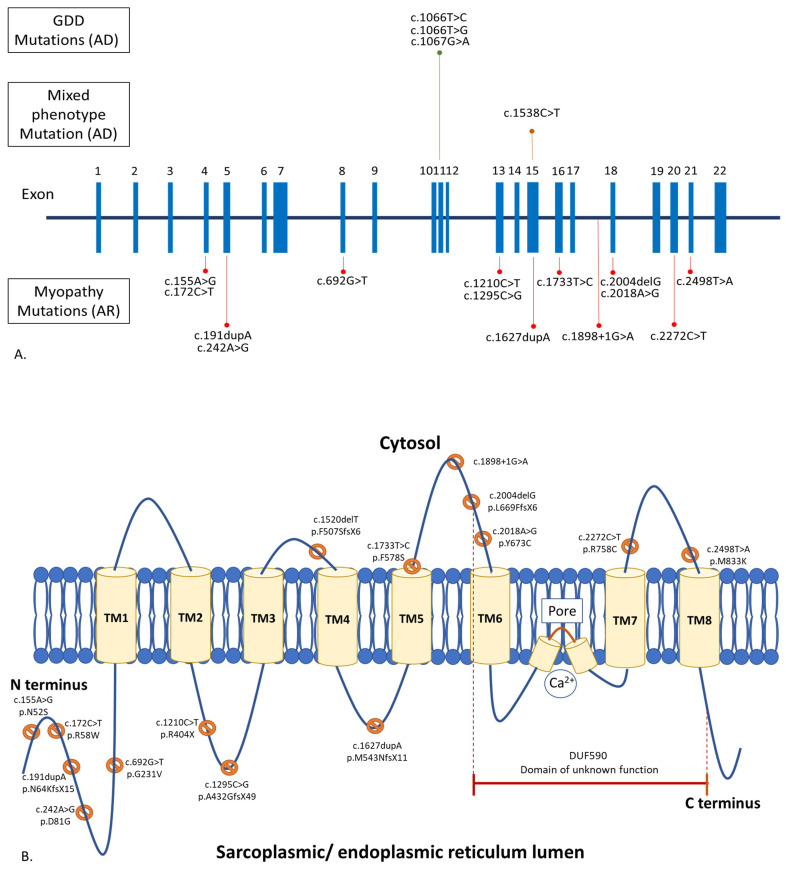
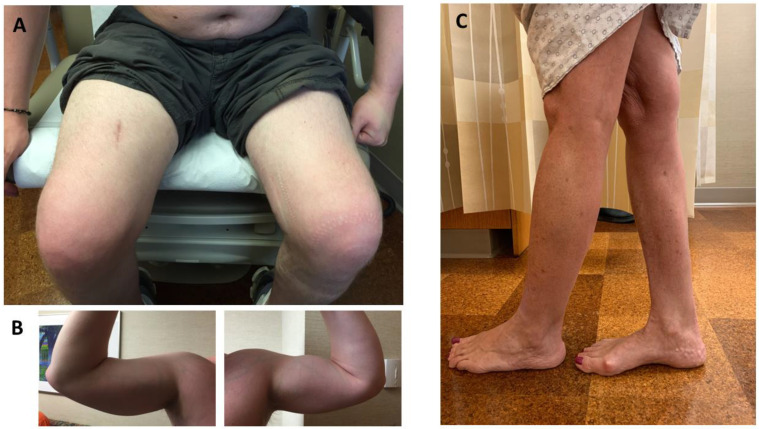
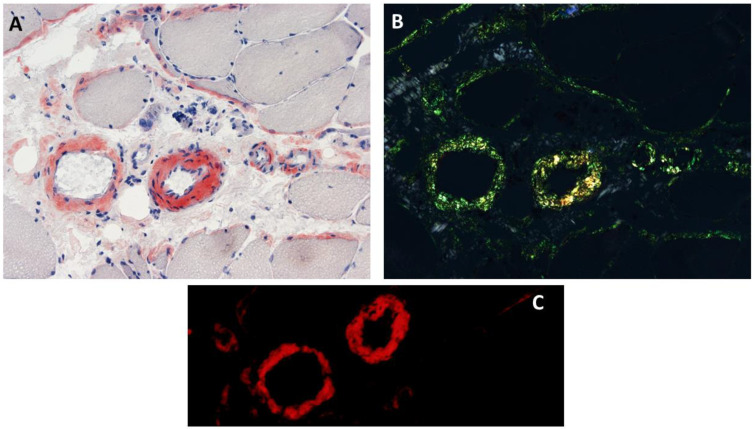
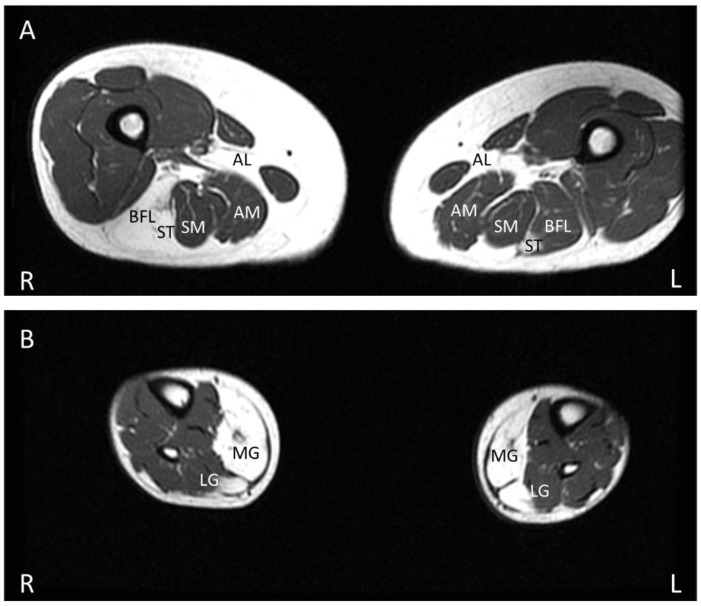
Anoctaminopathy-5 refers to a group of hereditary skeletal muscle or bone disorders due to mutations in the anoctamin 5 (ANO5)-encoding gene, ANO5. ANO5 is a 913-amino acid protein of the anoctamin family that functions predominantly in phospholipid scrambling and plays a key role in the sarcolemmal repairing process. Monoallelic mutations in ANO5 give rise to an autosomal dominant skeletal dysplastic syndrome (gnathodiaphyseal dysplasia or GDD), while its biallelic mutations underlie a continuum of four autosomal recessive muscle phenotypes: (1). limb-girdle muscular dystrophy type R12 (LGMDR12); (2). Miyoshi distal myopathy type 3 (MMD3); (3). metabolic myopathy-like (pseudometabolic) phenotype; (4). asymptomatic hyperCKemia. ANO5 muscle disorders are rare, but their prevalence is relatively high in northern European populations because of the founder mutation c.191dupA. Weakness is generally asymmetric and begins in proximal muscles in LGMDR12 and in distal muscles in MMD3. Patients with the pseudometabolic or asymptomatic hyperCKemia phenotype have no weakness, but conversion to the LGMDR12 or MMD3 phenotype may occur as the disease progresses. There is no clear genotype-phenotype correlation. Muscle biopsy displays a broad spectrum of pathology, ranging from normal to severe dystrophic changes. Intramuscular interstitial amyloid deposits are observed in approximately half of the patients. Symptomatic and supportive strategies remain the mainstay of treatment. The recent development of animal models of ANO5 muscle diseases could help achieve a better understanding of their underlying pathomechanisms and provide an invaluable resource for therapeutic discovery.
Keywords: ANO5; LGMD; LGMDR12; MMD3; Miyoshi myopathy; anoctamin 5; anoctaminopathy; limb–girdle muscular dystrophy.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Anoctamin-5 related muscle disease: clinical and genetic findings in a large European cohort.Brain. 2023 Sep 1;146(9):3800-3815. doi: 10.1093/brain/awad088. Brain. 2023. PMID: 36913258
-
Anoctamin 5 Knockout Mouse Model Recapitulates LGMD2L Muscle Pathology and Offers Insight Into in vivo Functional Deficits.J Neuromuscul Dis. 2021;8(s2):S243-S255. doi: 10.3233/JND-210720. J Neuromuscul Dis. 2021. PMID: 34633328 Free PMC article.
-
Phenotypic Spectrum of Myopathies with Recessive Anoctamin-5 Mutations.J Neuromuscul Dis. 2020;7(4):443-451. doi: 10.3233/JND-200515. J Neuromuscul Dis. 2020. PMID: 32925086
-
Single-centre experience with autosomal recessive limb-girdle muscular dystrophy: case series and literature review.Arq Neuropsiquiatr. 2023 Oct;81(10):922-933. doi: 10.1055/s-0043-1772833. Epub 2023 Oct 18. Arq Neuropsiquiatr. 2023. PMID: 37852290 Free PMC article. Review.
-
ANO5-related muscle diseases: From clinics and genetics to pathology and research strategies.Genes Dis. 2022 Feb 14;9(6):1506-1520. doi: 10.1016/j.gendis.2022.01.001. eCollection 2022 Nov. Genes Dis. 2022. PMID: 36157496 Free PMC article. Review.
Cited by
-
Novel Variant in ANO5 Muscular Dystrophy: Identification by Whole Genome Sequencing and Quad Analysis.Genes (Basel). 2024 Oct 6;15(10):1300. doi: 10.3390/genes15101300. Genes (Basel). 2024. PMID: 39457424 Free PMC article.
-
Hypertrophic Cardiomyopathy Complicated by Post-COVID-19 Myopericarditis in Patient with ANO5-Related Distal Myopathy.Genes (Basel). 2023 Jun 24;14(7):1332. doi: 10.3390/genes14071332. Genes (Basel). 2023. PMID: 37510237 Free PMC article.
-
Activation of thousands of genes in the lungs and kidneys by sepsis is countered by the selective nuclear blockade.Front Immunol. 2023 Aug 11;14:1221102. doi: 10.3389/fimmu.2023.1221102. eCollection 2023. Front Immunol. 2023. PMID: 37638006 Free PMC article.
-
Editorial for the Genetics of Muscular Dystrophies from the Pathogenesis to Gene Therapy Special Issue.Genes (Basel). 2023 Apr 17;14(4):926. doi: 10.3390/genes14040926. Genes (Basel). 2023. PMID: 37107684 Free PMC article.
References
-
- Tsutsumi S., Kamata N., Vokes T.J., Maruoka Y., Nakakuki K., Enomoto S., Omura K., Amagasa T., Nagayama M., Saito-Ohara F., et al. The novel gene encoding a putative transmembrane protein is mutated in gnathodiaphyseal dysplasia (GDD) Am. J. Hum. Genet. 2004;74:1255–1261. doi: 10.1086/421527. - DOI - PMC - PubMed
-
- Bolduc V., Marlow G., Boycott K.M., Saleki K., Inoue H., Kroon J., Itakura M., Robitaille Y., Parent L., Baas F., et al. Recessive mutations in the putative calcium-activated chloride channel Anoctamin 5 cause proximal LGMD2L and distal MMD3 muscular dystrophies. Am. J. Hum. Genet. 2010;86:213–221. doi: 10.1016/j.ajhg.2009.12.013. - DOI - PMC - PubMed
-
- Papadopoulos C., LaforÊt P., Nectoux J., Stojkovic T., Wahbi K., Carlier R.Y., Carlier P.G., Leonard-Louis S., Leturcq F., Romero N., et al. Hyperckemia and myalgia are common presentations of anoctamin-5-related myopathy in French patients. Muscle Nerve. 2017;56:1096–1100. doi: 10.1002/mus.25608. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
