

Novel mutation in coagulation factor VII (Carmel mutation): Identification and characterization
- PMID: 34027285
- PMCID: PMC8117812
- DOI: 10.1002/rth2.12407
Novel mutation in coagulation factor VII (Carmel mutation): Identification and characterization
Abstract
Background: Measurement of factor VII (FVII) activity does not enable prediction of bleeding tendency in individuals with inherited FVII deficiency.
Objective: To characterize the molecular and functional features of FVII in a family with FVII deficiency and correlate them with the bleeding tendency.
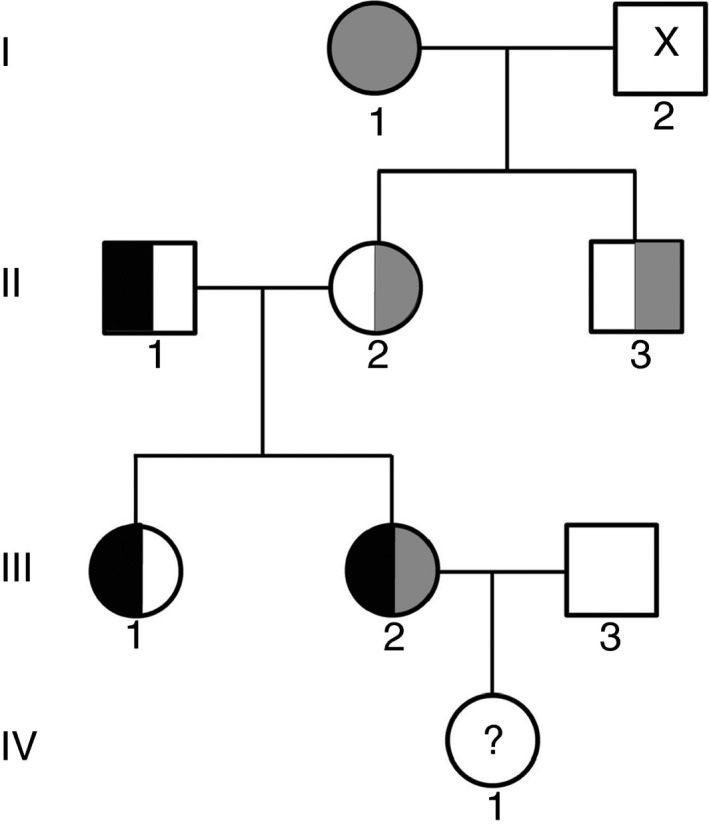
Patients/methods: We studied 7 family members with very low FVII activity using prothrombin time (PT), activated factor VII (FVIIa), FVII activity level, and thrombin generation. The factor 7 gene was sequenced and the mutation was analyzed by prediction software.
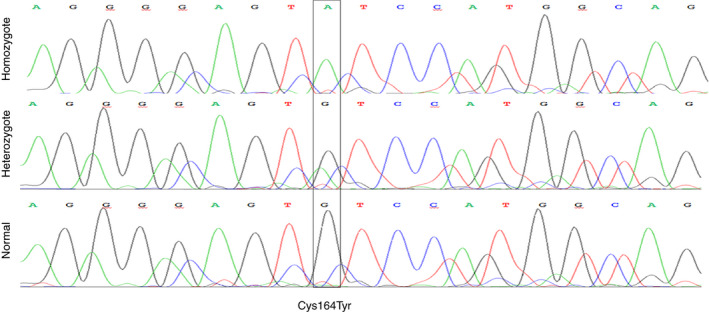
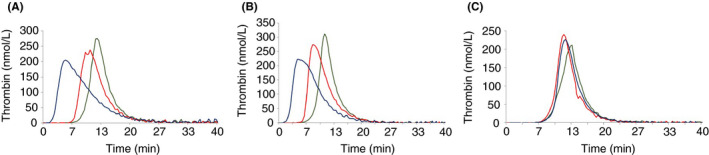
Results: The proband has very low FVII activity (0%-4%), with PT ranging between 5% to 18% depending on the tissue factor (TF) origin. Direct sequencing demonstrated a single homozygous nucleotide substitution G > A in exon 6, predicting a novel missense mutation Cys164Tyr. Three members of the family were found to be heterozygous carriers of this mutation. One of them was a compound heterozygote, carrying both the Cys164Tyr and Ala244Val mutation (linked to Arg353Gln polymorphism). Her FVII activity and antigen levels were 3%-7% and 8%, respectively. The other heterozygous carriers demonstrated FVII activity of 41%-54%, FVII antigen of 46%-66%, and FVIIa activity of 30%. FVIIa was undetectable in the homozygous and compound heterozygous subjects. Thrombin generation was normal in the presence of calcium, but no response to TF addition was observed in the homozygous proband, and a reduced response was observed in the compound heterozygous subject.
Conclusion: The patient homozygous for the "Carmel" mutation has mild clinical manifestations despite very low FVII activity, which correlates with thrombin generation results.
Keywords: bleeding disorders; factor VII; mutation; thrombin generation.
© 2021 The Authors. Research and Practice in Thrombosis and Haemostasis published by Wiley Periodicals LLC on behalf of International Society on Thrombosis and Haemostasis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

Similar articles
-
Analysis of Phenotype and Genotypein an Inherited Coagulation Factor VII Deficiency Pedigree.Clin Lab. 2019 Dec 1;65(12). doi: 10.7754/Clin.Lab.2019.190710. Clin Lab. 2019. PMID: 31850724
-
[Molecular analysis of two pedigrees with inherited coagulation factor VII deficiency].Zhonghua Er Ke Za Zhi. 2012 Nov;50(11):817-20. Zhonghua Er Ke Za Zhi. 2012. PMID: 23302610 Chinese.
-
[Genotype and phenotype analysis of congenital coagulator factor VII deficiency in four Chinese pedigrees].Zhonghua Xue Ye Xue Za Zhi. 2011 Mar;32(3):147-52. Zhonghua Xue Ye Xue Za Zhi. 2011. PMID: 21535950 Chinese.
-
The Story of Serum Prothrombin Conversion Accelerator, Proconvertin, Stable Factor, Cothromboplastin, Prothrombin Accelerator or Autoprothrombin I, and Their Subsequent Merging into Factor VII.Semin Thromb Hemost. 2015 Jun;41(4):366-73. doi: 10.1055/s-0035-1549851. Epub 2015 May 14. Semin Thromb Hemost. 2015. PMID: 25973586 Review.
-
Novel heterozygous F7 gene mutation (c. C1286T) associated with congenital factor VII deficiency: A case report and literature review.J Clin Lab Anal. 2022 May;36(5):e24349. doi: 10.1002/jcla.24349. Epub 2022 Mar 29. J Clin Lab Anal. 2022. PMID: 35349734 Free PMC article. Review.
References
-
- McVey JH, Boswell E, Mumford AD, Kemball‐Cook G, Tuddenham EG. Factor VII deficiency and the FVII mutation database. Hum Mutat. 2001;17:3–17. - PubMed
-
- Fromovich‐Amit Y, Zivelin A, Rosenberg N, Tamary H, Landau M. Seligsohn U. Characterization of mutations causing factor VII deficiency in 61 unrelated Israeli patients. J Thromb Haemost. 2004;2(10):1774–81. - PubMed
-
- Tamary H, Fromovich‐Amit Y, Shalmon L, Zaizov R, Yaniv I, Klar A, et al. Seligsohn U. Molecular characterization of four novel mutations causing factor VII deficiency. Hematol J. 2000;1(6):382–9. - PubMed
-
- Triplett DA, Brandt JT, Batard MA, Dixon JL, Fair DS. Hereditary factor VII deficiency: heterogeneity defined by combined functional and immunochemical analysis. Blood. 1985;66:1284–7. - PubMed
-
- Girolami A, Bertozzi I, de Marinis GB, Bonamigo E, Fabris F. Activated FVII levels in factor VII Padua (Arg304Gln) coagulation disorder and in true factor VII deficiency: a study in homozygotes and heterozygotes. Hematology. 2011;16(5):308–12. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
