

Two cases of carbonic anhydrase VA deficiency-An ultrarare metabolic decompensation syndrome presenting with hyperammonemia, lactic acidosis, ketonuria, and good clinical outcome
- PMID: 33473334
- PMCID: PMC7802620
- DOI: 10.1002/jmd2.12171
Two cases of carbonic anhydrase VA deficiency-An ultrarare metabolic decompensation syndrome presenting with hyperammonemia, lactic acidosis, ketonuria, and good clinical outcome
Abstract
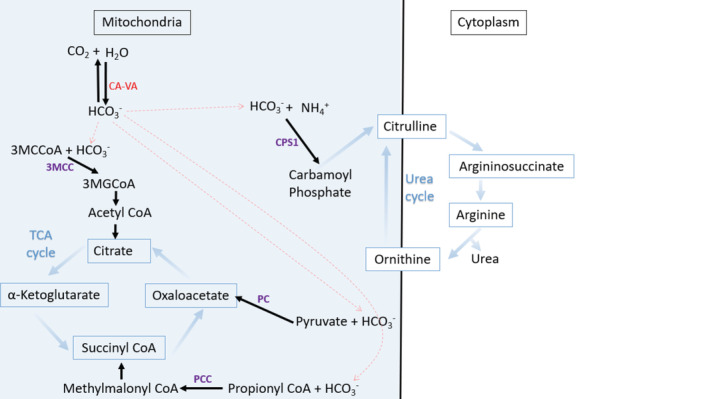
The combination of neonatal hyperammonemia, lactic acidosis, ketonuria, and hypoglycemia is pathognomonic for carbonic anhydrase VA (CA-VA) deficiency. We present two cases of this rare inborn error of metabolism. Both newborns with South Asian ancestry presented with a metabolic decompensation characterized by hyperammonemia, lactic acidosis and ketonuria; one also had hypoglycemia. Standard metabolic investigations (plasma amino acids, acylcarnitine profile, and urine organic acids) were not indicative of a specific organic aciduria or fatty acid oxidation defect but had some overlapping features with a urea cycle disorder (elevated glutamine, orotic acid, and low arginine). Hyperammonemia was treated initially with nitrogen scavenger therapy and carglumic acid. One patient required hemodialysis. Both have had a favorable long-term prognosis after their initial metabolic decompensation. Genetic testing confirmed the diagnosis of carbonic anhydrase VA (CA-VA) deficiency due to biallelic pathogenic variants in CA5A. These cases are in line with 15 cases previously described in the literature, making the phenotypic presentation pathognomonic for this ultrarare (potentially underdiagnosed) inborn error of metabolism with a good prognosis.
Keywords: encephalopathy; ketonuria; lactic acidosis; metabolic acidosis; neonatal hyperammonemia.
© 2020 The Authors. JIMD Reports published by John Wiley & Sons Ltd on behalf of SSIEM.
Conflict of interest statement
All authors have no conflict of interest to declare.
Figures
Similar articles
-
A founder mutation in CA5A causing intrafamilial and interfamilial phenotypic variability in a cohort of 18 patients with carbonic anhydrase VA deficiency.JIMD Rep. 2024 May 8;65(4):226-232. doi: 10.1002/jmd2.12426. eCollection 2024 Jul. JIMD Rep. 2024. PMID: 38974611 Free PMC article.
-
Carbonic anhydrase VA deficiency: a very rare case of hyperammonemic encephalopathy.J Pediatr Endocrinol Metab. 2020 Aug 18;33(10):1349-1352. doi: 10.1515/jpem-2020-0117. J Pediatr Endocrinol Metab. 2020. PMID: 32809955
-
Defective hepatic bicarbonate production due to carbonic anhydrase VA deficiency leads to early-onset life-threatening metabolic crisis.Genet Med. 2016 Oct;18(10):991-1000. doi: 10.1038/gim.2015.201. Epub 2016 Feb 25. Genet Med. 2016. PMID: 26913920
-
Impaired glucose homeostasis and a novel HLCS pathogenic variant in holocarboxylase synthetase deficiency: a report of two cases and brief review.J Pediatr Endocrinol Metab. 2020 Nov 26;33(11):1481-1486. doi: 10.1515/jpem-2020-0106. J Pediatr Endocrinol Metab. 2020. PMID: 32841162 Review.
-
Mitochondrial carbonic anhydrase VA and VB: properties and roles in health and disease.J Physiol. 2023 Jan;601(2):257-274. doi: 10.1113/JP283579. Epub 2022 Dec 19. J Physiol. 2023. PMID: 36464834 Free PMC article. Review.
Cited by
-
A founder mutation in CA5A causing intrafamilial and interfamilial phenotypic variability in a cohort of 18 patients with carbonic anhydrase VA deficiency.JIMD Rep. 2024 May 8;65(4):226-232. doi: 10.1002/jmd2.12426. eCollection 2024 Jul. JIMD Rep. 2024. PMID: 38974611 Free PMC article.
-
Recurrent hyperammonaemia in a patient with carbonic anhydrase VA deficiency.JIMD Rep. 2022 Aug 10;63(6):536-539. doi: 10.1002/jmd2.12322. eCollection 2022 Nov. JIMD Rep. 2022. PMID: 36341166 Free PMC article.
-
Hyperammonemia in Russia Due to Carbonic Anhydrase VA Deficiency Caused by Homozygous Mutation p.Lys185Lys (c.555G>A) of the CA5A Gene.Int J Mol Sci. 2022 Nov 30;23(23):15026. doi: 10.3390/ijms232315026. Int J Mol Sci. 2022. PMID: 36499355 Free PMC article.
-
A case of carbonic anhydrase type VA deficiency presenting as West syndrome in an infant with a novel mutation in the CA-VA gene.Epilepsy Behav Rep. 2022 Nov 7;20:100573. doi: 10.1016/j.ebr.2022.100573. eCollection 2022. Epilepsy Behav Rep. 2022. PMID: 36411877 Free PMC article.
-
An Extremely Rare Cause of Hyperammonemic Encephalopathy in an Infant.Indian J Pediatr. 2024 Jan;91(1):88. doi: 10.1007/s12098-023-04818-z. Epub 2023 Aug 23. Indian J Pediatr. 2024. PMID: 37610685 No abstract available.
References
-
- Diez‐Fernandez C, Rufenacht V, Santra S, et al. Defective hepatic bicarbonate production due to carbonic anhydrase VA deficiency leads to early‐onset life‐threatening metabolic crisis. Genet Med. 2016;18:991‐1000. - PubMed
-
- Baertling F, Wagner M, Brunet T, et al. Fatal metabolic decompensation in carbonic anhydrase VA deficiency despite early treatment and control of hyperammonemia. Genet Med. 2020;22:654‐655. - PubMed
-
- Haberle J. Carglumic acid for the treatment of N‐acetylglutamate synthase deficiency and acute hyperammonemia. Expert Rev Endocrinol Metab. 2012;7:263‐271. - PubMed
-
- Haberle J, Rubio V. Hyperammonemias and related disorders In: Blau N, Duran M, Gibson KM, Dionisi‐Vici C, eds. Physician's Guide to the Diagnosis, Treatment, and Follow‐Up of Inherited Metabolic Diseases. Heidelberg, New York, Dordrecht, London: Springer‐Verlag; 2014:47‐62.
Publication types
LinkOut - more resources
Full Text Sources
