

Molecular overview of progressive familial intrahepatic cholestasis
- PMID: 33384548
- PMCID: PMC7754551
- DOI: 10.3748/wjg.v26.i47.7470
Molecular overview of progressive familial intrahepatic cholestasis
Abstract
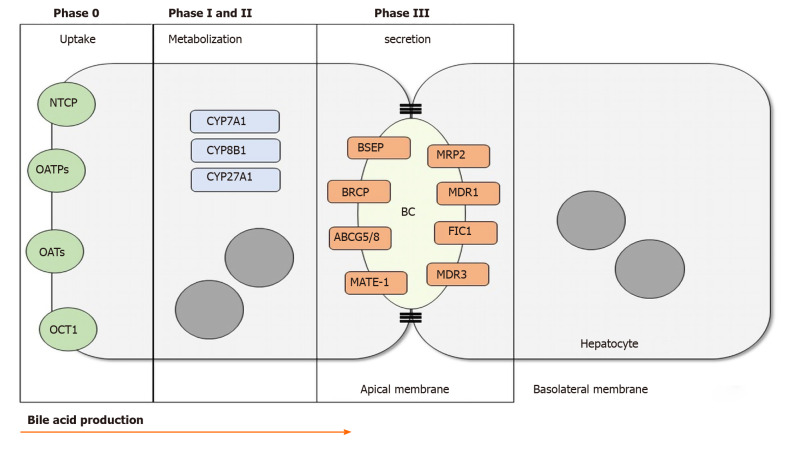
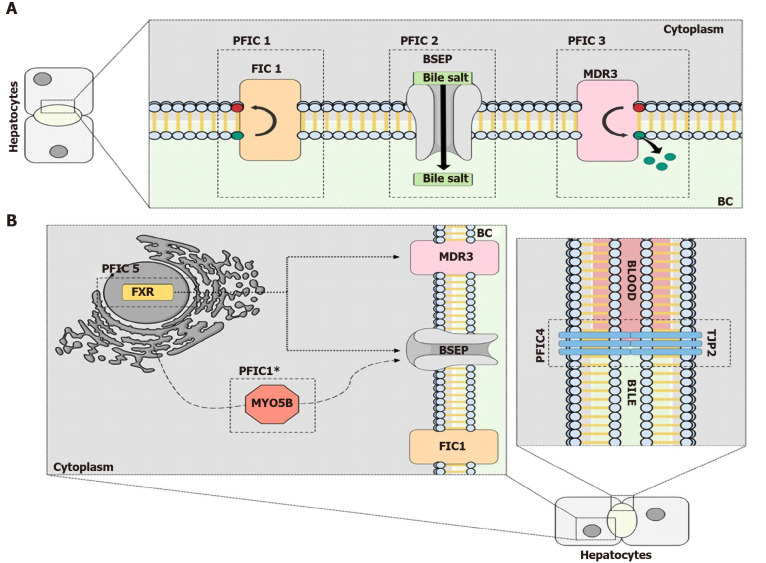
Cholestasis is a clinical condition resulting from the imapairment of bile flow. This condition could be caused by defects of the hepatocytes, which are responsible for the complex process of bile formation and secretion, and/or caused by defects in the secretory machinery of cholangiocytes. Several mutations and pathways that lead to cholestasis have been described. Progressive familial intrahepatic cholestasis (PFIC) is a group of rare diseases caused by autosomal recessive mutations in the genes that encode proteins expressed mainly in the apical membrane of the hepatocytes. PFIC 1, also known as Byler's disease, is caused by mutations of the ATP8B1 gene, which encodes the familial intrahepatic cholestasis 1 protein. PFIC 2 is characterized by the downregulation or absence of functional bile salt export pump (BSEP) expression via variations in the ABCB11 gene. Mutations of the ABCB4 gene result in lower expression of the multidrug resistance class 3 glycoprotein, leading to the third type of PFIC. Newer variations of this disease have been described. Loss of function of the tight junction protein 2 protein results in PFIC 4, while mutations of the NR1H4 gene, which encodes farnesoid X receptor, an important transcription factor for bile formation, cause PFIC 5. A recently described type of PFIC is associated with a mutation in the MYO5B gene, important for the trafficking of BSEP and hepatocyte membrane polarization. In this review, we provide a brief overview of the molecular mechanisms and clinical features associated with each type of PFIC based on peer reviewed journals published between 1993 and 2020.
Keywords: ABCB11/bile salt export pump; ABCB4/multidrug resistance class 3; ATP8B1/familial intrahepatic cholestasis 1; Bile; Intrahepatic cholestasis; Progressive familial intrahepatic cholestasis.
©The Author(s) 2020. Published by Baishideng Publishing Group Inc. All rights reserved.
Conflict of interest statement
Conflict-of-interest statement: A.S.-G., is co-founder and have a financial interest in Von Baer Wolff, Inc. a company focused on biofabrication of autologous human hepatocytes from stem cells technology and programming liver failure and their interests are managed by the Conflict of Interest Office at the University of Pittsburgh in accordance with their policies
Figures
Similar articles
-
Autoimmune BSEP disease: disease recurrence after liver transplantation for progressive familial intrahepatic cholestasis.Clin Rev Allergy Immunol. 2015 Jun;48(2-3):273-84. doi: 10.1007/s12016-014-8457-4. Clin Rev Allergy Immunol. 2015. PMID: 25342496 Review.
-
Progressive familial intrahepatic cholestasis.Clin Res Hepatol Gastroenterol. 2012 Sep;36 Suppl 1:S26-35. doi: 10.1016/S2210-7401(12)70018-9. Clin Res Hepatol Gastroenterol. 2012. PMID: 23141890 Review.
-
Newer variants of progressive familial intrahepatic cholestasis.World J Hepatol. 2021 Dec 27;13(12):2024-2038. doi: 10.4254/wjh.v13.i12.2024. World J Hepatol. 2021. PMID: 35070006 Free PMC article. Review.
-
NR1H4 analysis in patients with progressive familial intrahepatic cholestasis, drug-induced cholestasis or intrahepatic cholestasis of pregnancy unrelated to ATP8B1, ABCB11 and ABCB4 mutations.Clin Res Hepatol Gastroenterol. 2012 Dec;36(6):569-73. doi: 10.1016/j.clinre.2012.08.008. Epub 2012 Nov 9. Clin Res Hepatol Gastroenterol. 2012. PMID: 23142591
-
ATP8B1 and ABCB11 analysis in 62 children with normal gamma-glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): phenotypic differences between PFIC1 and PFIC2 and natural history.Hepatology. 2010 May;51(5):1645-55. doi: 10.1002/hep.23539. Hepatology. 2010. PMID: 20232290
Cited by
-
Biallelic Novel USP53 Splicing Variant Disrupting the Gene Function that Causes Cholestasis Phenotype and Review of the Literature.Mol Syndromol. 2023 Jan;13(6):471-484. doi: 10.1159/000523937. Epub 2022 Apr 28. Mol Syndromol. 2023. PMID: 36660033 Free PMC article.
-
Genetic alterations and molecular mechanisms underlying hereditary intrahepatic cholestasis.Front Pharmacol. 2023 May 31;14:1173542. doi: 10.3389/fphar.2023.1173542. eCollection 2023. Front Pharmacol. 2023. PMID: 37324459 Free PMC article. Review.
-
Association of Very Rare NOTCH2 Variants with Clinical Features of Alagille Syndrome.Genes (Basel). 2024 Aug 6;15(8):1034. doi: 10.3390/genes15081034. Genes (Basel). 2024. PMID: 39202394 Free PMC article.
-
Network pharmacology analysis and molecular docking to unveil the potential mechanisms of San-Huang-Chai-Zhu formula treating cholestasis.PLoS One. 2022 Feb 23;17(2):e0264398. doi: 10.1371/journal.pone.0264398. eCollection 2022. PLoS One. 2022. PMID: 35196362 Free PMC article.
-
Odevixibat: a promising new treatment for progressive familial intrahepatic cholestasis.Expert Opin Pharmacother. 2022 Nov;23(16):1771-1779. doi: 10.1080/14656566.2022.2140040. Epub 2022 Oct 30. Expert Opin Pharmacother. 2022. PMID: 36278881 Free PMC article.
References
-
- Hirschfield GM, Heathcote EJ, Gershwin ME. Pathogenesis of cholestatic liver disease and therapeutic approaches. Gastroenterology. 2010;139:1481–1496. - PubMed
-
- Alvarez L, Jara P, Sánchez-Sabaté E, Hierro L, Larrauri J, Díaz MC, Camarena C, De la Vega A, Frauca E, López-Collazo E, Lapunzina P. Reduced hepatic expression of farnesoid X receptor in hereditary cholestasis associated to mutation in ATP8B1. Hum Mol Genet. 2004;13:2451–2460. - PubMed
-
- Gomez-Ospina N, Potter CJ, Xiao R, Manickam K, Kim MS, Kim KH, Shneider BL, Picarsic JL, Jacobson TA, Zhang J, He W, Liu P, Knisely AS, Finegold MJ, Muzny DM, Boerwinkle E, Lupski JR, Plon SE, Gibbs RA, Eng CM, Yang Y, Washington GC, Porteus MH, Berquist WE, Kambham N, Singh RJ, Xia F, Enns GM, Moore DD. Mutations in the nuclear bile acid receptor FXR cause progressive familial intrahepatic cholestasis. Nat Commun. 2016;7:10713. - PMC - PubMed
-
- Marson FAL, Bertuzzo CS, Ribeiro JD. Classification of CFTR mutation classes. Lancet Respir Med. 2016;4:e37–e38. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
