

Biallelic variants in KYNU cause a multisystemic syndrome with hand hyperphalangism
- PMID: 31923704
- PMCID: PMC10521254
- DOI: 10.1016/j.bone.2019.115219
Biallelic variants in KYNU cause a multisystemic syndrome with hand hyperphalangism
Abstract
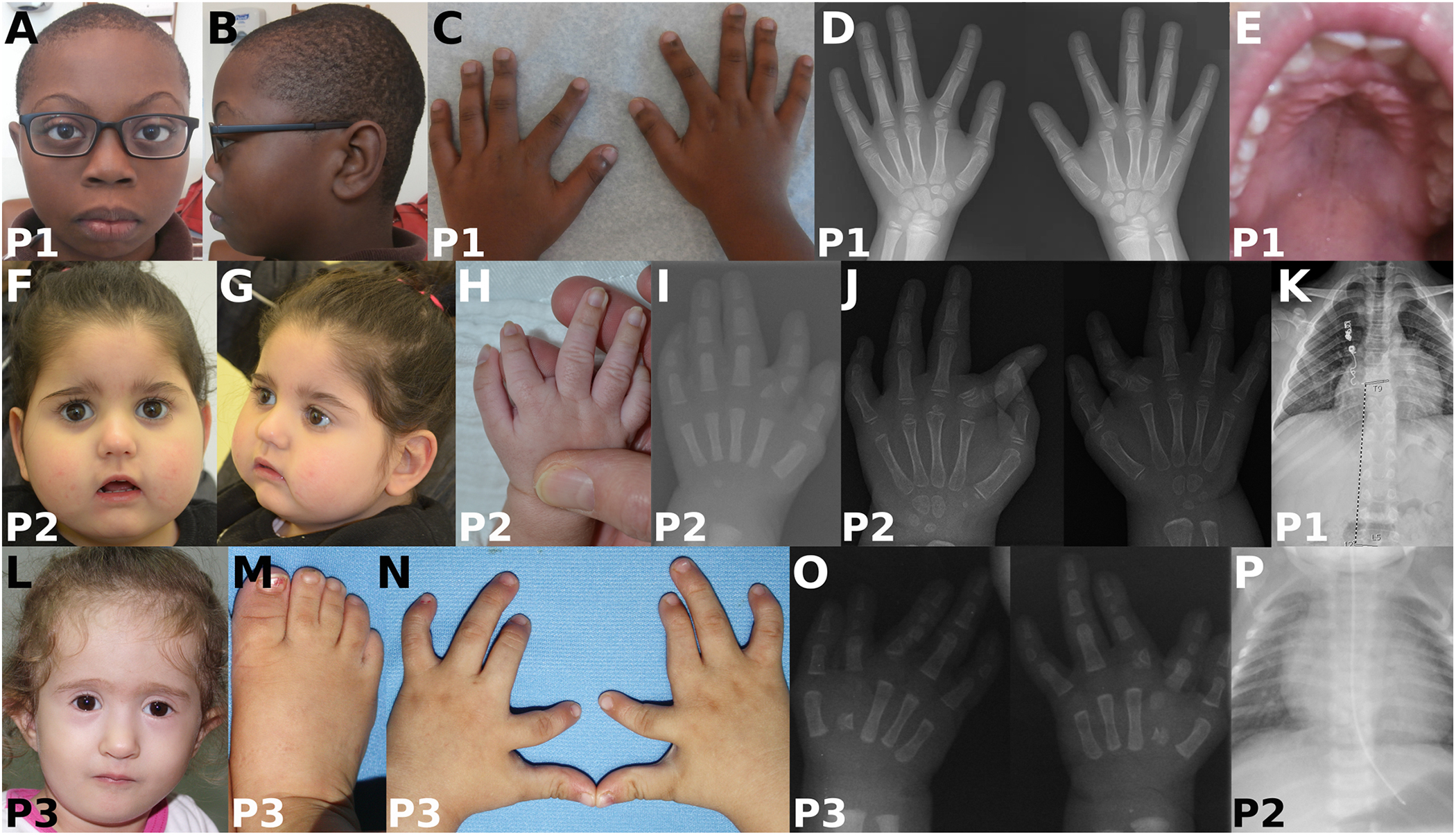
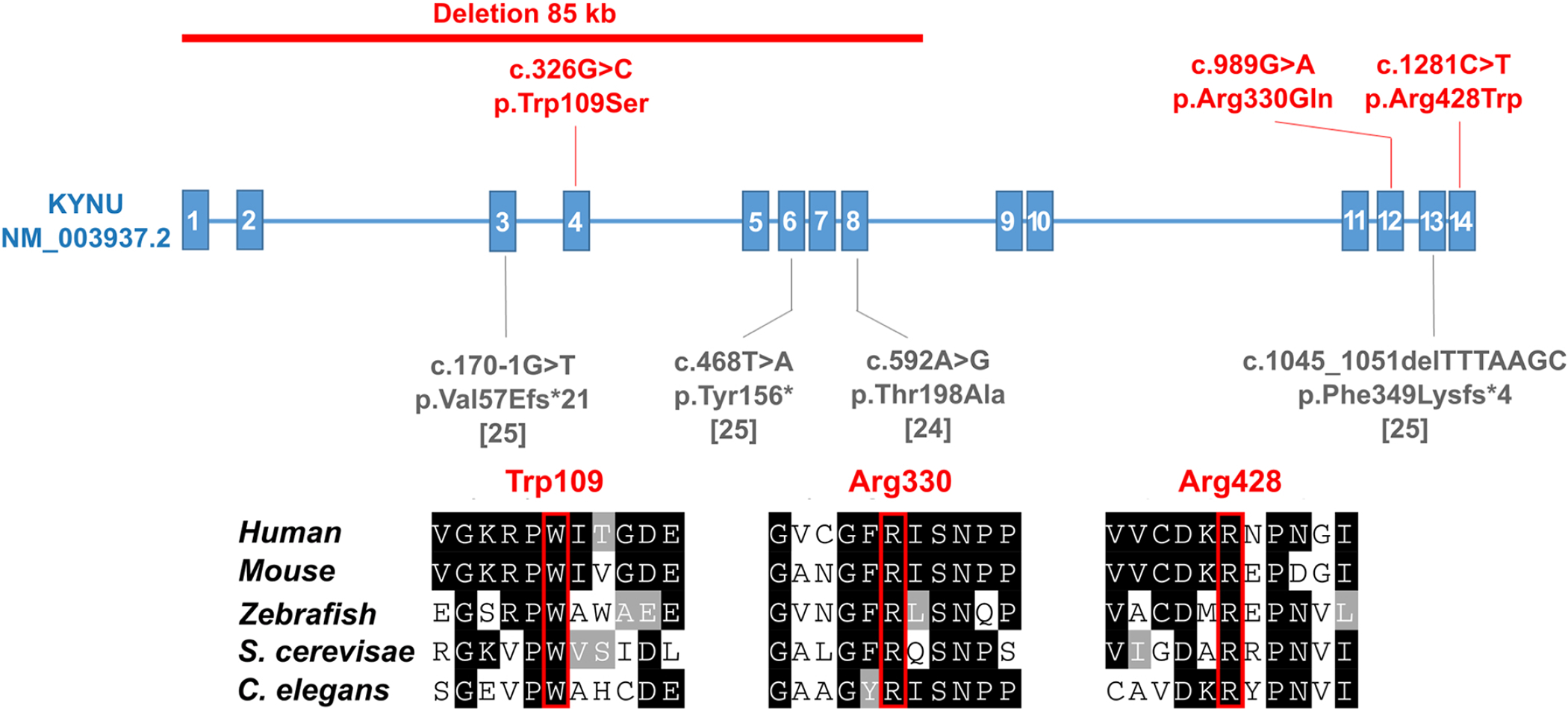
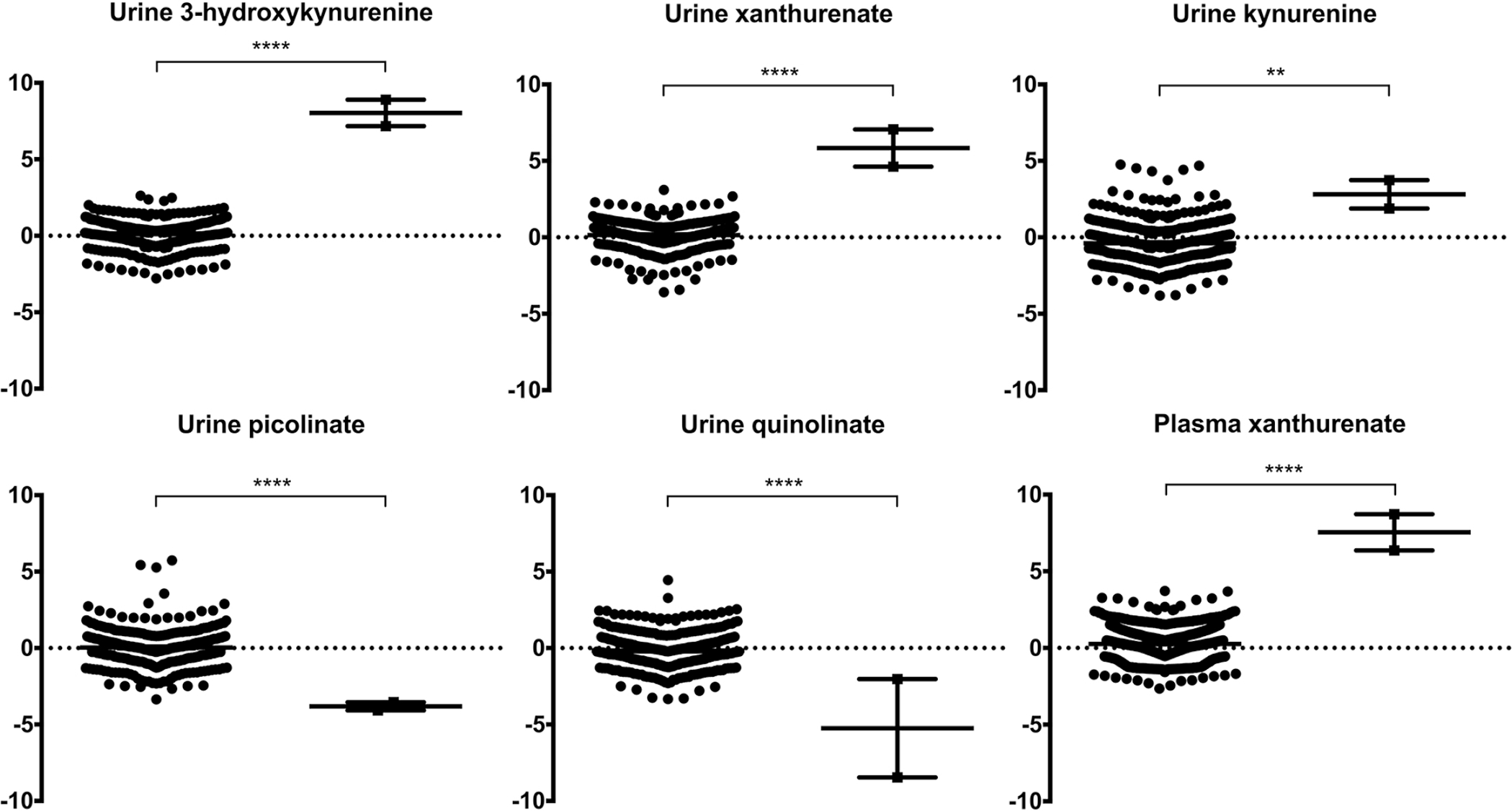
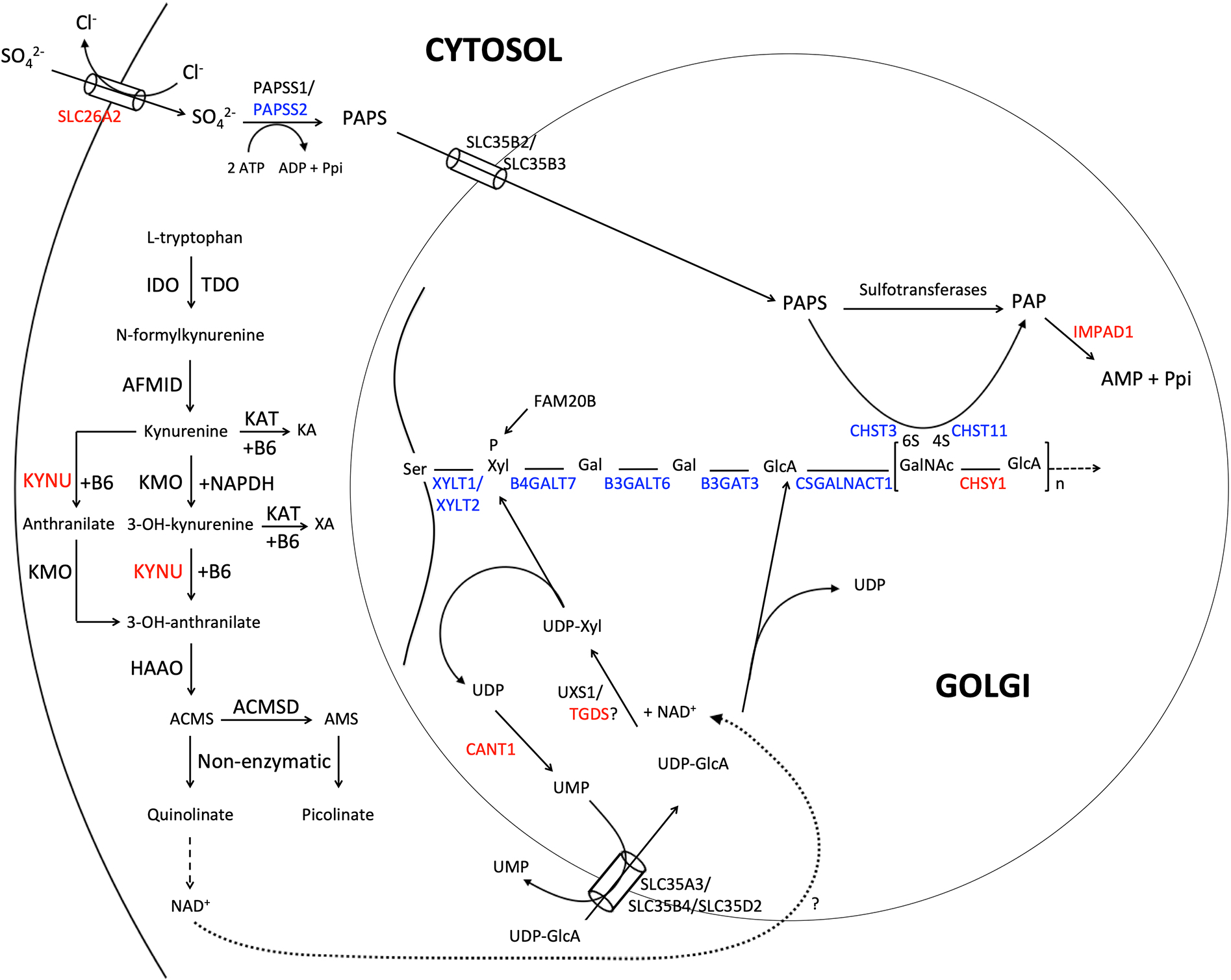
Catel-Manzke syndrome is characterized by the combination of Pierre Robin sequence and radial deviation, shortening as well as clinodactyly of the index fingers, due to an accessory ossification center. Mutations in TGDS have been identified as one cause of Catel-Manzke syndrome, but cannot be found as causative in every patient with the clinical diagnosis. We performed a chromosome microarray and/or exome sequencing in three patients with hand hyperphalangism, heart defect, short stature, and mild to severe developmental delay, all of whom were initially given a clinical diagnosis of Catel-Manzke syndrome. In one patient, we detected a large deletion of exons 1-8 and the missense variant c.1282C > T (p.Arg428Trp) in KYNU (NM_003937.2), whereas homozygous missense variants in KYNU were found in the other two patients (c.989G > A (p.Arg330Gln) and c.326G > C (p.Trp109Ser)). Plasma and urine metabolomic analysis of two patients indicated a block along the tryptophan catabolic pathway and urine organic acid analysis showed excretion of xanthurenic acid. Biallelic loss-of-function mutations in KYNU were recently described as a cause of NAD deficiency with vertebral, cardiac, renal and limb defects; however, no hand hyperphalangism was described in those patients, and Catel-Manzke syndrome was not discussed as a differential diagnosis. In conclusion, we present unrelated patients identified with biallelic variants in KYNU leading to kynureninase deficiency and xanthurenic aciduria as a very likely cause of their hyperphalangism, heart defect, short stature, and developmental delay. We suggest performance of urine organic acid analysis in patients with suspected Catel-Manzke syndrome, particularly in those with cardiac or vertebral defects or without mutations in TGDS.
Keywords: Catel-Manzke syndrome; Hyperphalangism; KYNU; Kynureninase deficiency; Skeletal dysplasia; Xanthurenic aciduria.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of competing interest Claudia Gonzaga-Jauregui is a full-time employee of the Regeneron Genetics Center from Regeneron Pharmaceuticals Inc. and receives stock options as part of compensation. All other authors declare that they have no conflict of interest.
Figures
Similar articles
-
A Homozygous Deletion of Exon 5 of KYNU Resulting from a Maternal Chromosome 2 Isodisomy (UPD2) Causes Catel-Manzke-Syndrome/VCRL Syndrome.Genes (Basel). 2021 Jun 7;12(6):879. doi: 10.3390/genes12060879. Genes (Basel). 2021. PMID: 34200361 Free PMC article.
-
TGDS pathogenic variants cause Catel-Manzke syndrome without hyperphalangy.Am J Med Genet A. 2020 Mar;182(3):431-436. doi: 10.1002/ajmg.a.61419. Epub 2019 Nov 25. Am J Med Genet A. 2020. PMID: 31769200
-
Mutations in TGDS associated with additional malformations of the middle fingers and halluces: Atypical Catel-Manzke syndrome in a fetus.Am J Med Genet A. 2017 Jun;173(6):1694-1697. doi: 10.1002/ajmg.a.38209. Epub 2017 Apr 19. Am J Med Genet A. 2017. PMID: 28422407
-
Homozygous loss of function BRCA1 variant causing a Fanconi-anemia-like phenotype, a clinical report and review of previous patients.Eur J Med Genet. 2018 Mar;61(3):130-133. doi: 10.1016/j.ejmg.2017.11.003. Epub 2017 Nov 10. Eur J Med Genet. 2018. PMID: 29133208 Review.
-
A review of craniofacial disorders caused by spliceosomal defects.Clin Genet. 2015 Nov;88(5):405-15. doi: 10.1111/cge.12596. Epub 2015 May 1. Clin Genet. 2015. PMID: 25865758 Review.
Cited by
-
NAD+ deficiency in human congenital malformations and miscarriage: A new model of pleiotropy.Am J Med Genet A. 2022 Sep;188(9):2834-2849. doi: 10.1002/ajmg.a.62764. Epub 2022 Apr 29. Am J Med Genet A. 2022. PMID: 35484986 Free PMC article.
-
Supplementation with NAD+ and its precursors: A rescue of female reproductive diseases.Biochem Biophys Rep. 2024 Apr 23;38:101715. doi: 10.1016/j.bbrep.2024.101715. eCollection 2024 Jul. Biochem Biophys Rep. 2024. PMID: 38698835 Free PMC article. Review.
-
A Homozygous Deletion of Exon 5 of KYNU Resulting from a Maternal Chromosome 2 Isodisomy (UPD2) Causes Catel-Manzke-Syndrome/VCRL Syndrome.Genes (Basel). 2021 Jun 7;12(6):879. doi: 10.3390/genes12060879. Genes (Basel). 2021. PMID: 34200361 Free PMC article.
-
New cases that expand the genotypic and phenotypic spectrum of Congenital NAD Deficiency Disorder.Hum Mutat. 2021 Jul;42(7):862-876. doi: 10.1002/humu.24211. Epub 2021 May 16. Hum Mutat. 2021. PMID: 33942433 Free PMC article.
-
A metabolic signature for NADSYN1-dependent congenital NAD deficiency disorder.J Clin Invest. 2024 Feb 15;134(4):e174824. doi: 10.1172/JCI174824. J Clin Invest. 2024. PMID: 38357931 Free PMC article.
References
-
- Catel W, Differentialdiagnose von Krankheitssymptomen bei Kindern und Jugendlichen, 3 ed., Stuttgart: G. Thieme1961.
-
- Manzke H, [Symmetrical hyperphalangy of the second finger by a supplementary metacarpus bone], Fortschritte auf dem Gebiete der Rontgenstrahlen und der Nuklearmedizin 105(3) (1966) 425–7. - PubMed
-
- Manzke H, Lehmann K, Klopocki E, Caliebe A, Catel-Manzke syndrome: two new patients and a critical review of the literature, European journal of medical genetics 51(5) (2008) 452–65. - PubMed
-
- Ehmke N, Caliebe A, Koenig R, Kant SG, Stark Z, Cormier-Daire V, Wieczorek D, Gillessen-Kaesbach G, Hoff K, Kawalia A, Thiele H, Altmuller J, Fischer-Zirnsak B, Knaus A, Zhu N, Heinrich V, Huber C, Harabula I, Spielmann M, Horn D, Kornak U, Hecht J, Krawitz PM, Nurnberg P, Siebert R, Manzke H, Mundlos S, Homozygous and compound-heterozygous mutations in TGDS cause Catel-Manzke syndrome, American journal of human genetics 95(6) (2014) 763–70. - PMC - PubMed
-
- Schoner K, Bald R, Horn D, Rehder H, Kornak U, Ehmke N, Mutations in TGDS associated with additional malformations of the middle fingers and halluces: Atypical Catel-Manzke syndrome in a fetus, American journal of medical genetics. Part A 173(6) (2017) 1694–1697. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
