

Jaundice revisited: recent advances in the diagnosis and treatment of inherited cholestatic liver diseases
- PMID: 30367658
- PMCID: PMC6203212
- DOI: 10.1186/s12929-018-0475-8
Jaundice revisited: recent advances in the diagnosis and treatment of inherited cholestatic liver diseases
Abstract
Background: Jaundice is a common symptom of inherited or acquired liver diseases or a manifestation of diseases involving red blood cell metabolism. Recent progress has elucidated the molecular mechanisms of bile metabolism, hepatocellular transport, bile ductular development, intestinal bile salt reabsorption, and the regulation of bile acids homeostasis.
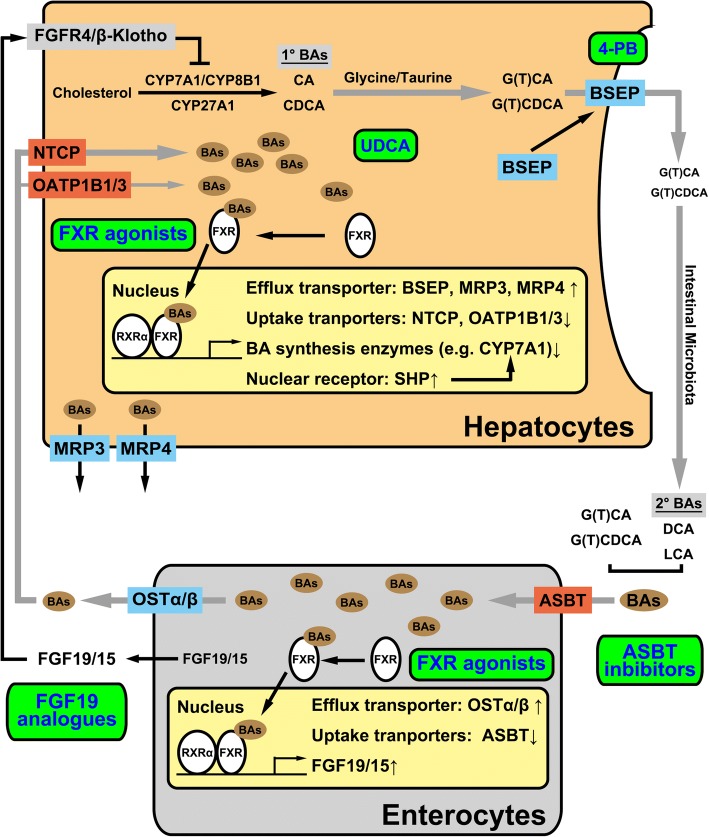
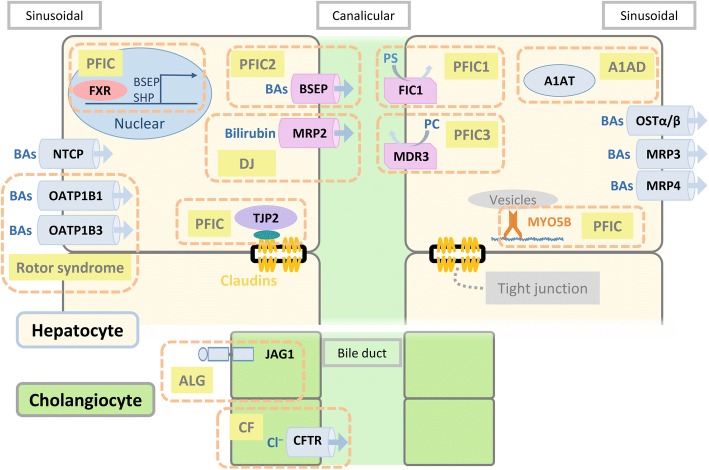
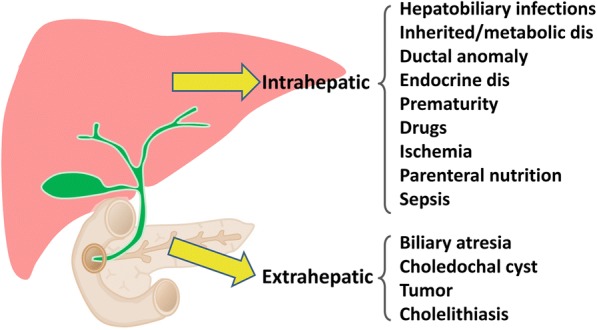
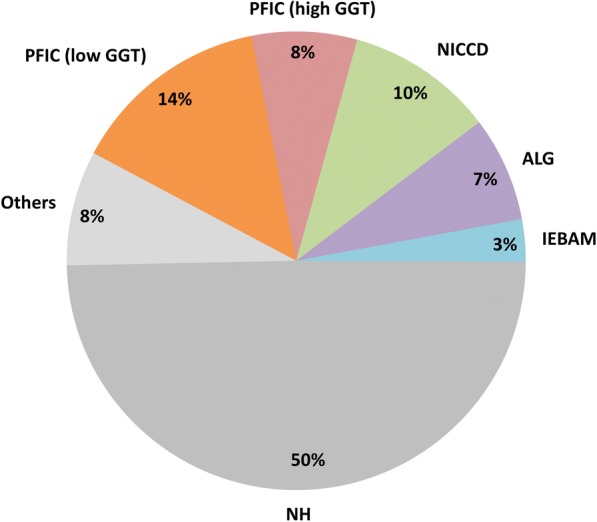
Main body: The major genetic diseases causing jaundice involve disturbances of bile flow. The insufficiency of bile salts in the intestines leads to fat malabsorption and fat-soluble vitamin deficiencies. Accumulation of excessive bile acids and aberrant metabolites results in hepatocellular injury and biliary cirrhosis. Progressive familial intrahepatic cholestasis (PFIC) is the prototype of genetic liver diseases manifesting jaundice in early childhood, progressive liver fibrosis/cirrhosis, and failure to thrive. The first three types of PFICs identified (PFIC1, PFIC2, and PFIC3) represent defects in FIC1 (ATP8B1), BSEP (ABCB11), or MDR3 (ABCB4). In the last 5 years, new genetic disorders, such as TJP2, FXR, and MYO5B defects, have been demonstrated to cause a similar PFIC phenotype. Inborn errors of bile acid metabolism also cause progressive cholestatic liver injuries. Prompt differential diagnosis is important because oral primary bile acid replacement may effectively reverse liver failure and restore liver functions. DCDC2 is a newly identified genetic disorder causing neonatal sclerosing cholangitis. Other cholestatic genetic disorders may have extra-hepatic manifestations, such as developmental disorders causing ductal plate malformation (Alagille syndrome, polycystic liver/kidney diseases), mitochondrial hepatopathy, and endocrine or chromosomal disorders. The diagnosis of genetic liver diseases has evolved from direct sequencing of a single gene to panel-based next generation sequencing. Whole exome sequencing and whole genome sequencing have been actively investigated in research and clinical studies. Current treatment modalities include medical treatment (ursodeoxycholic acid, cholic acid or chenodeoxycholic acid), surgery (partial biliary diversion and liver transplantation), symptomatic treatment for pruritus, and nutritional therapy. New drug development based on gene-specific treatments, such as apical sodium-dependent bile acid transporter (ASBT) inhibitor, for BSEP defects are underway.
Short conclusion: Understanding the complex pathways of jaundice and cholestasis not only enhance insights into liver pathophysiology but also elucidate many causes of genetic liver diseases and promote the development of novel treatments.
Keywords: Bile acids; Cholestasis; Genetic liver disease; Next generation sequencing; Pediatric; Progressive familial intrahepatic cholestasis.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable (the manuscript does not content patient information).
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
Similar articles
-
Progressive familial intrahepatic cholestasis.Clin Res Hepatol Gastroenterol. 2012 Sep;36 Suppl 1:S26-35. doi: 10.1016/S2210-7401(12)70018-9. Clin Res Hepatol Gastroenterol. 2012. PMID: 23141890 Review.
-
Progressive familial intrahepatic cholestasis.Orphanet J Rare Dis. 2009 Jan 8;4:1. doi: 10.1186/1750-1172-4-1. Orphanet J Rare Dis. 2009. PMID: 19133130 Free PMC article. Review.
-
Progressive familial intrahepatic cholestasis.J Clin Exp Hepatol. 2014 Mar;4(1):25-36. doi: 10.1016/j.jceh.2013.10.005. Epub 2013 Nov 23. J Clin Exp Hepatol. 2014. PMID: 25755532 Free PMC article. Review.
-
ATP8B1 and ABCB11 analysis in 62 children with normal gamma-glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): phenotypic differences between PFIC1 and PFIC2 and natural history.Hepatology. 2010 May;51(5):1645-55. doi: 10.1002/hep.23539. Hepatology. 2010. PMID: 20232290
-
Progressive familial intrahepatic cholestasis and inborn errors of bile acid synthesis.Clin Res Hepatol Gastroenterol. 2012 Jun;36(3):271-4. doi: 10.1016/j.clinre.2012.03.020. Epub 2012 May 18. Clin Res Hepatol Gastroenterol. 2012. PMID: 22609295
Cited by
-
Acinar ATP8b1/LPC pathway promotes macrophage efferocytosis and clearance of inflammation during chronic pancreatitis development.Cell Death Dis. 2022 Oct 22;13(10):893. doi: 10.1038/s41419-022-05322-6. Cell Death Dis. 2022. PMID: 36273194 Free PMC article.
-
The ESCRT-III molecules regulate the apical targeting of bile salt export pump.J Biomed Sci. 2021 Mar 9;28(1):19. doi: 10.1186/s12929-020-00706-2. J Biomed Sci. 2021. PMID: 33750401 Free PMC article.
-
Clinical, pathological and genetic characteristics of 17 unrelated children with Alagille Syndrome.BMC Pediatr. 2024 Aug 20;24(1):532. doi: 10.1186/s12887-024-04973-y. BMC Pediatr. 2024. PMID: 39164659 Free PMC article.
-
Clinical and Laboratory Characteristics in Children with Alagille Syndrome: Experience of a Single Center.Int J Gen Med. 2023 Jan 6;16:77-83. doi: 10.2147/IJGM.S382430. eCollection 2023. Int J Gen Med. 2023. PMID: 36636710 Free PMC article.
-
Fraxinellone Induces Hepatotoxicity in Zebrafish through Oxidative Stress and the Transporters Pathway.Molecules. 2022 Apr 20;27(9):2647. doi: 10.3390/molecules27092647. Molecules. 2022. PMID: 35566003 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
