

The ten-year evolutionary trajectory of a highly recurrent paediatric high grade neuroepithelial tumour with MN1:BEND2 fusion
- PMID: 29348602
- PMCID: PMC5773598
- DOI: 10.1038/s41598-018-19389-9
The ten-year evolutionary trajectory of a highly recurrent paediatric high grade neuroepithelial tumour with MN1:BEND2 fusion
Abstract
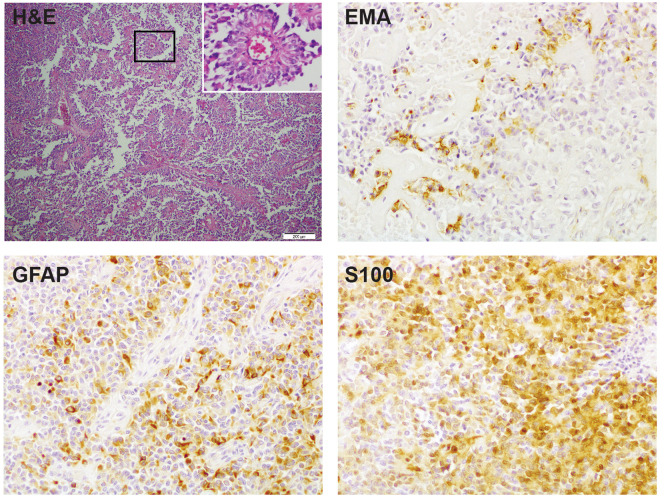
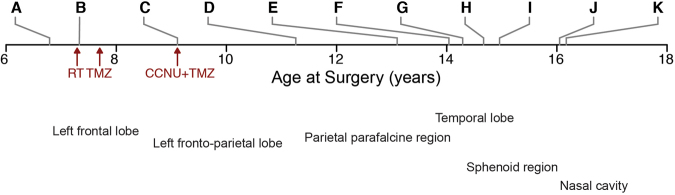
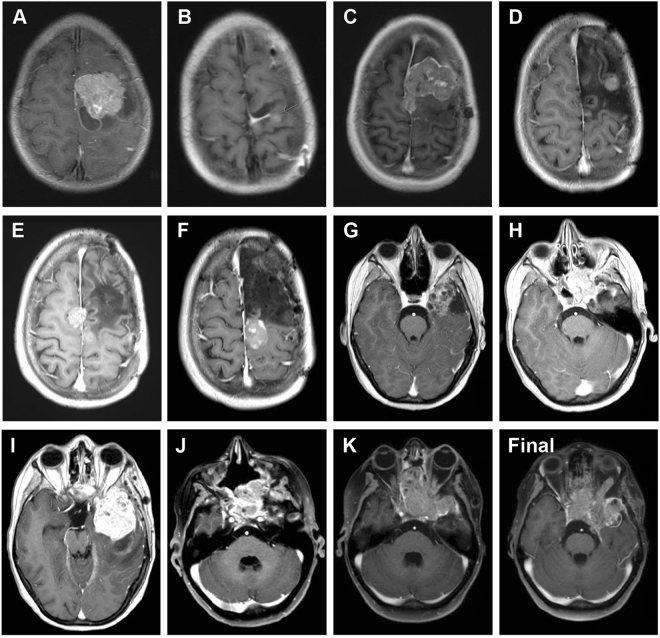
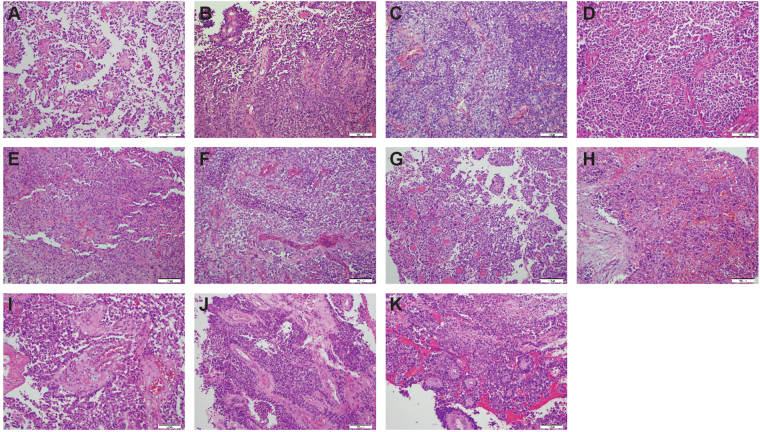
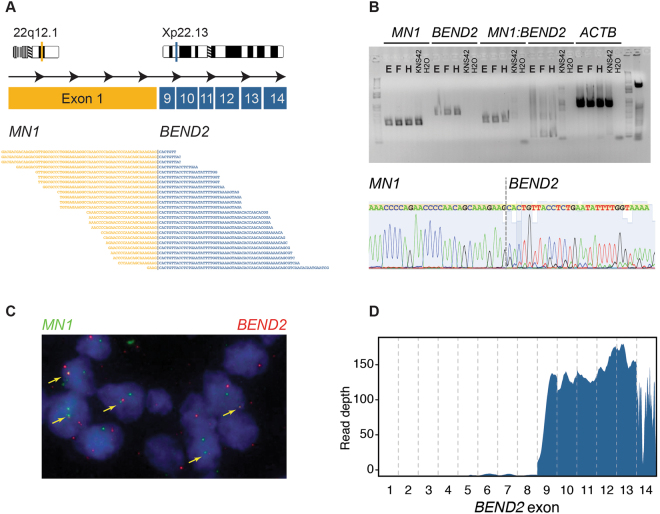
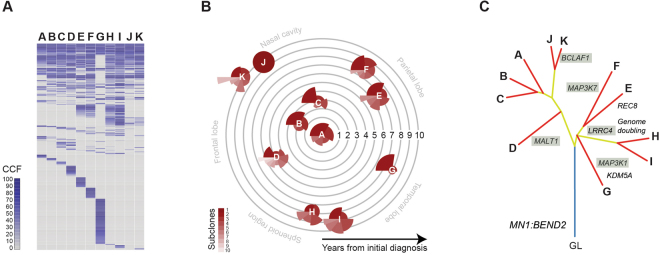
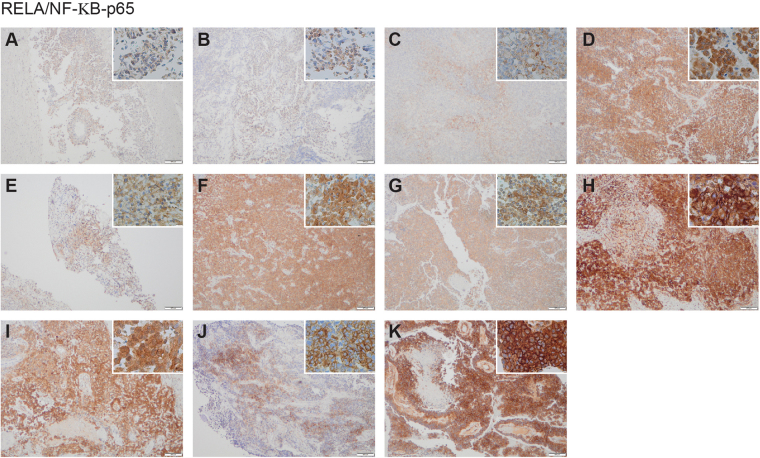
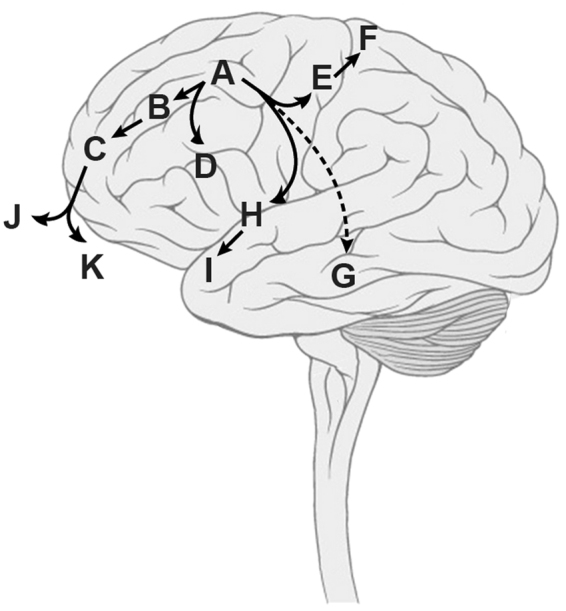
Astroblastomas are rare brain tumours which predominate in children and young adults, and have a controversial claim as a distinct entity, with no established WHO grade. Reports suggest a better outcome than high grade gliomas, though they frequently recur. Recently, they have been described to overlap with a newly-discovered group of tumours described as'high grade neuroepithelial tumour with MN1 alteration' (CNS HGNET-MN1), defined by global methylation patterns and strongly associated with gene fusions targeting MN1. We have studied a unique case of astroblastoma arising in a 6 year-old girl, with multiple recurrences over a period of 10 years, with the pathognomonic MN1:BEND2 fusion. Exome sequencing allowed for a phylogenetic reconstruction of tumour evolution, which when integrated with clinical, pathological and radiological data provide for a detailed understanding of disease progression, with initial treatment driving tumour dissemination along four distinct trajectories. Infiltration of distant sites was associated with a later genome doubling, whilst there was evidence of convergent evolution of different lesions acquiring distinct alterations targeting NF-κB. These data represent an unusual opportunity to understand the evolutionary history of a highly recurrent childhood brain tumour, and provide novel therapeutic targets for astroblastoma/CNS HGNET-MN1.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Early ependymal tumor with MN1-BEND2 fusion: a mostly cerebral tumor of female children with a good prognosis that is distinct from classical astroblastoma.J Neurooncol. 2023 Feb;161(3):425-439. doi: 10.1007/s11060-022-04222-1. Epub 2023 Jan 6. J Neurooncol. 2023. PMID: 36604386 Free PMC article. Review.
-
Spinal cord astroblastoma with EWSR1-BEND2 fusion classified as HGNET-MN1 by methylation classification: a case report.Brain Tumor Pathol. 2021 Oct;38(4):283-289. doi: 10.1007/s10014-021-00412-3. Epub 2021 Jul 27. Brain Tumor Pathol. 2021. PMID: 34313881
-
Paediatric astroblastoma-like neuroepithelial tumour of the spinal cord with a MAMLD1-BEND2 rearrangement.Neuropathol Appl Neurobiol. 2022 Aug;48(5):e12814. doi: 10.1111/nan.12814. Epub 2022 Apr 10. Neuropathol Appl Neurobiol. 2022. PMID: 35301744
-
Multimodal molecular analysis of astroblastoma enables reclassification of most cases into more specific molecular entities.Brain Pathol. 2018 Mar;28(2):192-202. doi: 10.1111/bpa.12561. Epub 2017 Oct 27. Brain Pathol. 2018. PMID: 28960623 Free PMC article.
-
MN1 rearrangement in astroblastoma: study of eight cases and review of literature.Brain Tumor Pathol. 2019 Jul;36(3):112-120. doi: 10.1007/s10014-019-00346-x. Epub 2019 May 20. Brain Tumor Pathol. 2019. PMID: 31111274 Review.
Cited by
-
Gene fusions are frequent in ACTH-secreting neuroendocrine neoplasms of the pancreas, but not in their non-pancreatic counterparts.Virchows Arch. 2023 Mar;482(3):507-516. doi: 10.1007/s00428-022-03484-4. Epub 2023 Jan 24. Virchows Arch. 2023. PMID: 36690805 Free PMC article.
-
A Low-grade Sinonasal Sarcoma Harboring EWSR1::BEND2: Expanding the Differential Diagnosis of Sinonasal Spindle Cell Neoplasms.Head Neck Pathol. 2023 Jun;17(2):571-575. doi: 10.1007/s12105-023-01527-z. Epub 2023 Jan 16. Head Neck Pathol. 2023. PMID: 36646985 Free PMC article.
-
Heterogeneous clinicopathological findings and patient-reported outcomes in adults with MN1-altered CNS tumors: A case report and systematic literature review.Front Oncol. 2023 Jan 19;13:1099618. doi: 10.3389/fonc.2023.1099618. eCollection 2023. Front Oncol. 2023. PMID: 36741001 Free PMC article.
-
Molecular identification of CNS NB-FOXR2, CNS EFT-CIC, CNS HGNET-MN1 and CNS HGNET-BCOR pediatric brain tumors using tumor-specific signature genes.Acta Neuropathol Commun. 2020 Jul 10;8(1):105. doi: 10.1186/s40478-020-00984-9. Acta Neuropathol Commun. 2020. PMID: 32650833 Free PMC article.
-
Early ependymal tumor with MN1-BEND2 fusion: a mostly cerebral tumor of female children with a good prognosis that is distinct from classical astroblastoma.J Neurooncol. 2023 Feb;161(3):425-439. doi: 10.1007/s11060-022-04222-1. Epub 2023 Jan 6. J Neurooncol. 2023. PMID: 36604386 Free PMC article. Review.
References
-
- Mangano, F. T., Bradford, A. C., Mittler, M. A., Valderrama, E. & Schneider, S. J. Astroblastoma. Case report, review of the literature, and analysis of treatment strategies. J Neurosurg Sci51, 21–27; discussion 27 (2007). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
