

Functional characterizations of rare UBA1 variants in X-linked Spinal Muscular Atrophy
- PMID: 29034082
- PMCID: PMC5615770
- DOI: 10.12688/f1000research.11878.1
Functional characterizations of rare UBA1 variants in X-linked Spinal Muscular Atrophy
Abstract
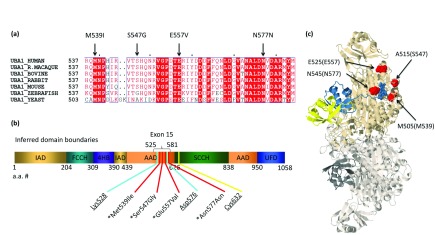
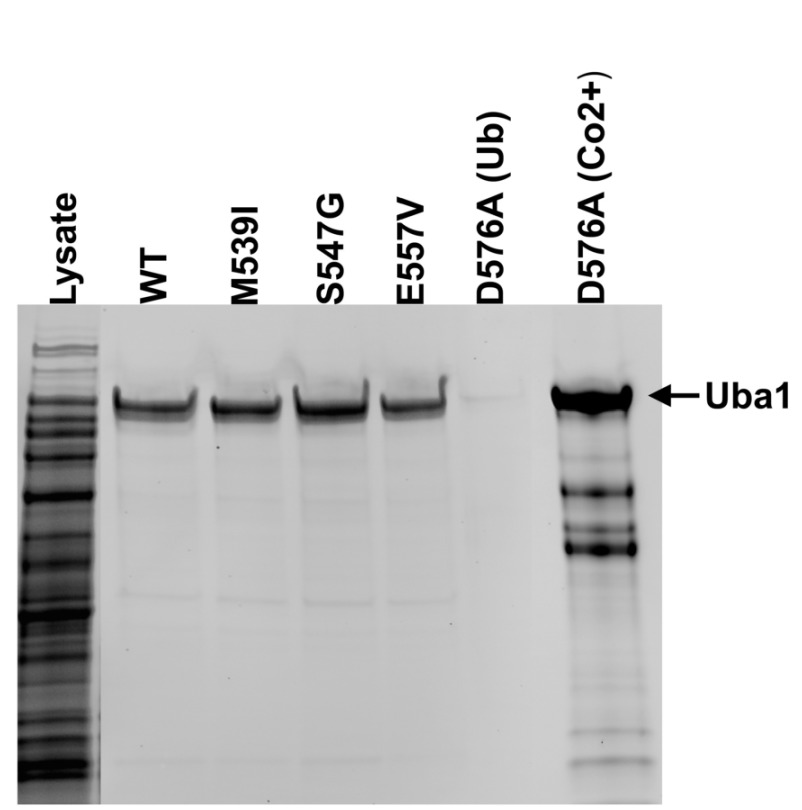
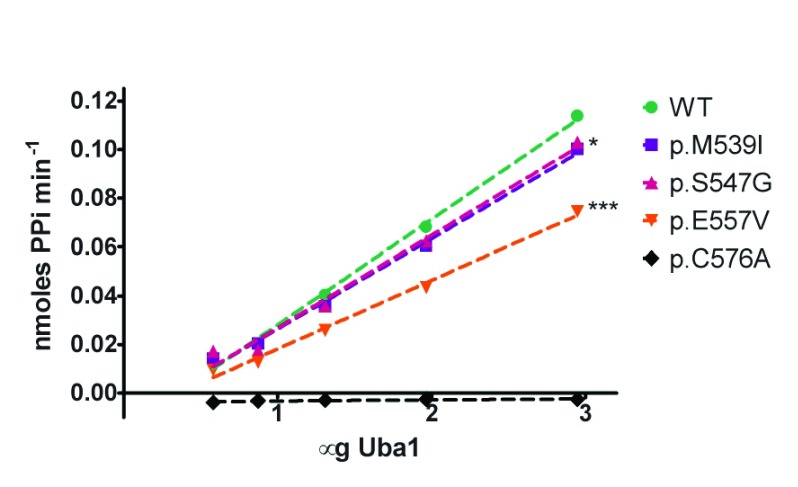
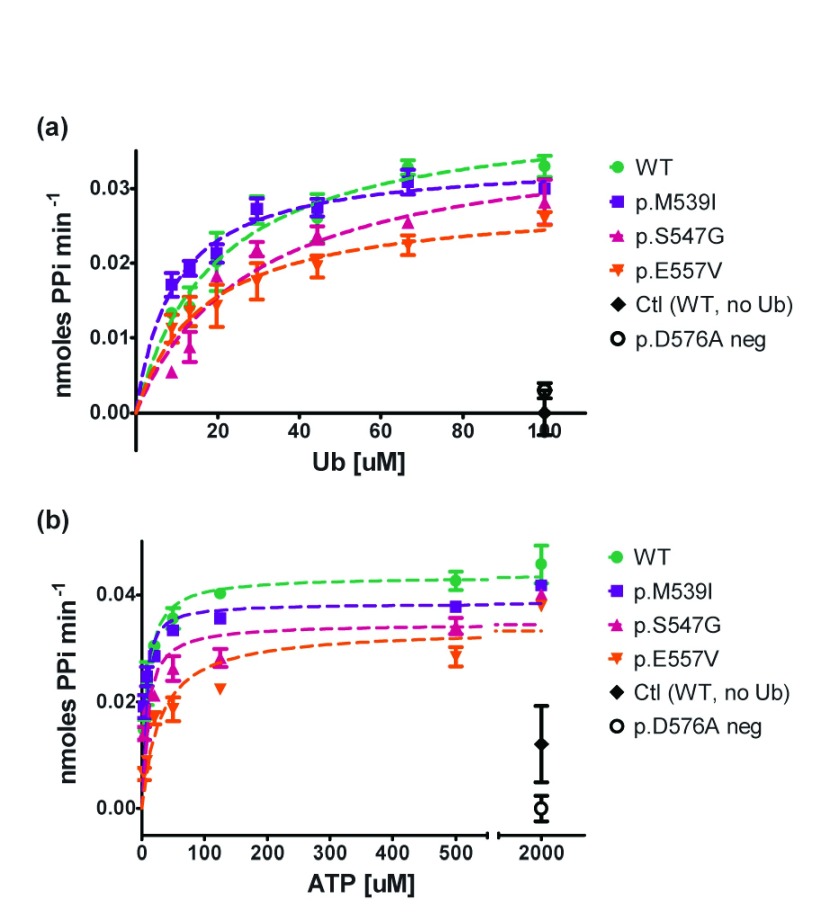
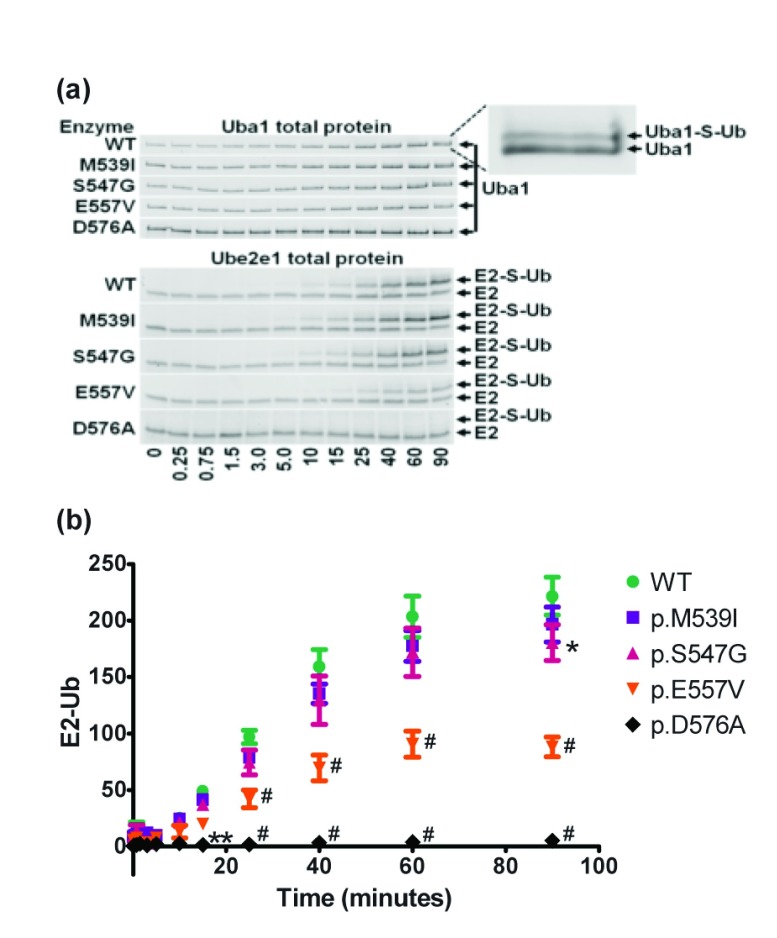
Background: X-linked spinal muscular atrophy (XL-SMA) results from mutations in the Ubiquitin-Like Modifier Activating Enzyme 1 ( UBA1). Previously, four novel closely clustered mutations have been shown to cause this fatal infantile disorder affecting only males. These mutations, three missense and one synonymous, all lie within Exon15 of the UBA1 gene, which contains the active adenylation domain (AAD). Methods: In this study, our group characterized the three known missense variants in vitro. Using a novel Uba1 assay and other methods, we investigated Uba1 adenylation, thioester, and transthioesterification reactions in vitro to determine possible biochemical effects of the missense variants. Results: Our data revealed that only one of the three XL-SMA missense variants impairs the Ubiquitin-adenylating ability of Uba1. Additionally, these missense variants retained Ubiquitin thioester bond formation and transthioesterification rates equal to that found in the wild type. Conclusions: Our results demonstrate a surprising shift from the likelihood of these XL-SMA mutations playing a damaging role in Uba1's enzymatic activity with Ubiquitin, to other roles such as altering UBA1 mRNA splicing via the disruption of splicing factor binding sites, similar to a mechanism in traditional SMA, or disrupting binding to other important in vivo binding partners. These findings help to narrow the search for the areas of possible dysfunction in the Ubiquitin-proteasome pathway that ultimately result in XL-SMA. Moreover, this investigation provides additional critical understanding of the mutations' biochemical mechanisms, vital for the development of future effective diagnostic assays and therapeutics.
Keywords: SMAX2; UBA1; X-linked spinal muscular atrophy; disease mechanisms; ubiquitin proteasome system; ubiquitination.
Conflict of interest statement
Competing interests: No competing interests were disclosed.
Figures
Similar articles
-
A novel Xp11.22-22.33 deletion suggesting a possible mechanism of congenital cervical spinal muscular atrophy.Mol Genet Genomic Med. 2021 Mar;9(3):e1606. doi: 10.1002/mgg3.1606. Epub 2021 Jan 29. Mol Genet Genomic Med. 2021. PMID: 33513289 Free PMC article.
-
A Pathogenic Missense Variant (c.1617G>A, p.Met539Ile) in UBA1 Causing Infantile X-Linked Spinal Muscular Atrophy (SMAX2).Front Pediatr. 2020 Feb 28;8:64. doi: 10.3389/fped.2020.00064. eCollection 2020. Front Pediatr. 2020. PMID: 32181232 Free PMC article.
-
Identification of UBA1 as the causative gene of an X-linked non-Kennedy spinal-bulbar muscular atrophy.Eur J Neurol. 2022 Dec;29(12):3556-3563. doi: 10.1111/ene.15528. Epub 2022 Sep 13. Eur J Neurol. 2022. PMID: 35996994
-
UBA1: At the Crossroads of Ubiquitin Homeostasis and Neurodegeneration.Trends Mol Med. 2015 Oct;21(10):622-632. doi: 10.1016/j.molmed.2015.08.003. Trends Mol Med. 2015. PMID: 26432019 Free PMC article. Review.
-
The pivotal role of ubiquitin-activating enzyme E1 (UBA1) in neuronal health and neurodegeneration.Int J Biochem Cell Biol. 2020 Jun;123:105746. doi: 10.1016/j.biocel.2020.105746. Epub 2020 Apr 18. Int J Biochem Cell Biol. 2020. PMID: 32315770 Review.
Cited by
-
A novel Xp11.22-22.33 deletion suggesting a possible mechanism of congenital cervical spinal muscular atrophy.Mol Genet Genomic Med. 2021 Mar;9(3):e1606. doi: 10.1002/mgg3.1606. Epub 2021 Jan 29. Mol Genet Genomic Med. 2021. PMID: 33513289 Free PMC article.
-
Shared and distinct mechanisms of UBA1 inactivation across different diseases.EMBO J. 2024 May;43(10):1919-1946. doi: 10.1038/s44318-024-00046-z. Epub 2024 Feb 15. EMBO J. 2024. PMID: 38360993 Free PMC article.
-
Disorders of ubiquitylation: unchained inflammation.Nat Rev Rheumatol. 2022 Aug;18(8):435-447. doi: 10.1038/s41584-022-00778-4. Epub 2022 May 6. Nat Rev Rheumatol. 2022. PMID: 35523963 Free PMC article. Review.
-
A Pathogenic Missense Variant (c.1617G>A, p.Met539Ile) in UBA1 Causing Infantile X-Linked Spinal Muscular Atrophy (SMAX2).Front Pediatr. 2020 Feb 28;8:64. doi: 10.3389/fped.2020.00064. eCollection 2020. Front Pediatr. 2020. PMID: 32181232 Free PMC article.
-
Auranofin targets UBA1 and enhances UBA1 activity by facilitating ubiquitin trans-thioesterification to E2 ubiquitin-conjugating enzymes.Nat Commun. 2023 Aug 9;14(1):4798. doi: 10.1038/s41467-023-40537-x. Nat Commun. 2023. PMID: 37558718 Free PMC article.
References
-
- Balak CD, Hunter JM, Ahearn ME, et al. : Dataset 1 in: Functional characterizations of rare UBA1 variants in X-linked Spinal Muscular Atrophy. F1000Research. 2017a. Data Source - DOI - PMC - PubMed
-
- Balak CD, Hunter JM, Ahearn ME, et al. : Dataset 2 in: Functional characterizations of rare UBA1 variants in X-linked Spinal Muscular Atrophy. F1000Research. 2017b. Data Source - DOI - PMC - PubMed
-
- Balak CD, Hunter JM, Ahearn ME, et al. : Dataset 3 in: Functional characterizations of rare UBA1 variants in X-linked Spinal Muscular Atrophy. F1000Research. 2017c. Data Source - DOI - PMC - PubMed
-
- Balak CD, Hunter JM, Ahearn ME, et al. : Dataset 4 in: Functional characterizations of rare UBA1 variants in X-linked Spinal Muscular Atrophy. F1000Research. 2017d. Data Source - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous