

The obligatory role of Activin A in the formation of heterotopic bone in Fibrodysplasia Ossificans Progressiva
- PMID: 28629737
- PMCID: PMC6706059
- DOI: 10.1016/j.bone.2017.06.011
The obligatory role of Activin A in the formation of heterotopic bone in Fibrodysplasia Ossificans Progressiva
Abstract
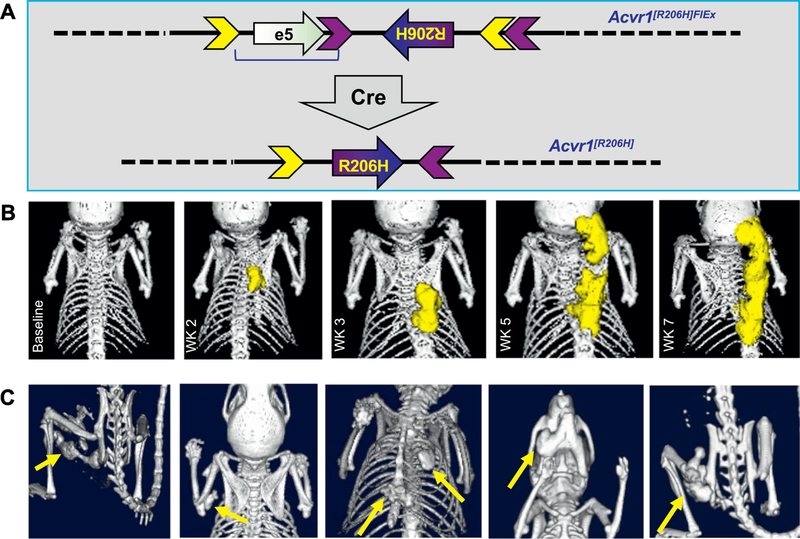
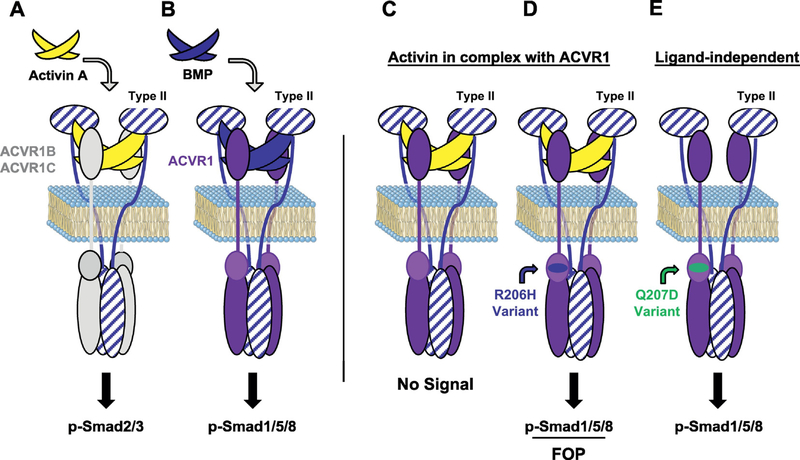
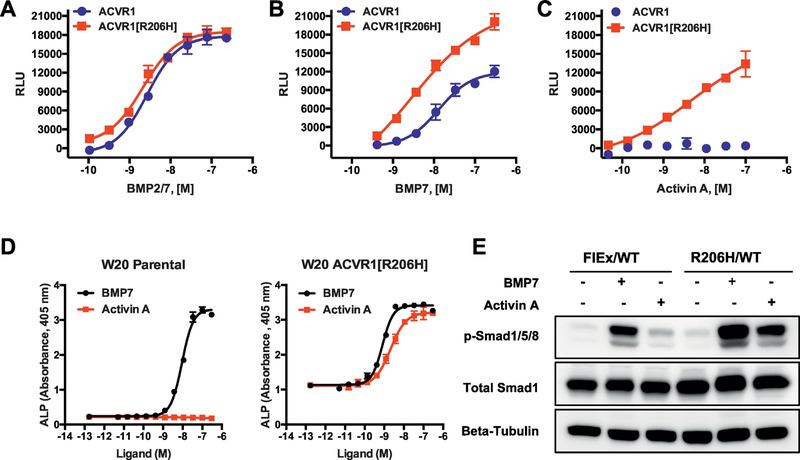
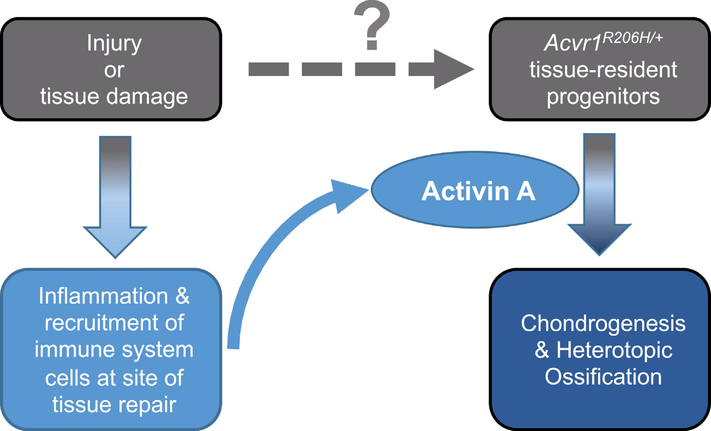
Fibrodysplasia Ossificans Progressiva (FOP) is a rare genetic disorder that presents at birth with only minor patterning defects, but manifests its debilitating pathology early in life with episodic, yet progressive and cumulative, heterotopic ossification (HO) of ligaments, tendons, and a subset of major skeletal muscles. The resulting HO lesions are endochondral in nature, and appear to be linked to inflammatory stimuli arising in association with known injuries, or from inflammation linked to normal tissue repair. FOP is caused by gain-of-function mutations in ACVR1, which encodes a type I BMP receptor. Initial studies on the pathogenic mechanism of FOP-causing mutations in ACVR1 focused on the enhanced function of this receptor in response to certain BMP ligands, or independently of ligands, but did not directly address the fact that HO in FOP is episodic and inflammation-driven. Recently, we and others demonstrated that Activin A is an obligate factor for the initiation of HO in FOP, signaling aberrantly via mutant ACVR1 to transduce osteogenic signals and trigger heterotopic bone formation (Hatsell et al., 2015; Hino et al., 2015). Subsequently, we identified distinct tissue-resident mesenchymal progenitor cells residing in muscles and tendons that recognize Activin A as a pro-osteogenic signal (solely in the context of FOP-causing mutant ACVR1), and give rise to the cartilaginous anlagen that form heterotopic bone (Dey et al., 2016). During the course of these studies, we also found that the activity of FOP-causing ACVR1 mutations does not by itself explain the triggered or inflammatory nature of HO in FOP, suggesting the importance of other, inflammation-introduced, factors or processes. This review presents a synthesis of these findings with a focus on the role of Activin A and inflammation in HO, and lays out perspectives for future research.
Keywords: ACVR1; Activin A; Anti-Activin antibody; Fibrodysplasia Ossificans Progressiva; Heterotopic Ossification; Progenitor cells.
Copyright © 2017 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
The ACVR1 R206H mutation found in fibrodysplasia ossificans progressiva increases human induced pluripotent stem cell-derived endothelial cell formation and collagen production through BMP-mediated SMAD1/5/8 signaling.Stem Cell Res Ther. 2016 Aug 17;7(1):115. doi: 10.1186/s13287-016-0372-6. Stem Cell Res Ther. 2016. PMID: 27530160 Free PMC article.
-
Overexpression of Wild-Type ACVR1 in Fibrodysplasia Ossificans Progressiva Mice Rescues Perinatal Lethality and Inhibits Heterotopic Ossification.J Bone Miner Res. 2022 Nov;37(11):2077-2093. doi: 10.1002/jbmr.4617. Epub 2022 Jul 3. J Bone Miner Res. 2022. PMID: 35637634 Free PMC article.
-
Anti-ACVR1 antibodies exacerbate heterotopic ossification in fibrodysplasia ossificans progressiva (FOP) by activating FOP-mutant ACVR1.J Clin Invest. 2022 Jun 15;132(12):e153792. doi: 10.1172/JCI153792. J Clin Invest. 2022. PMID: 35511419 Free PMC article.
-
The role of Activin A in fibrodysplasia ossificans progressiva: a prominent mediator.Biosci Rep. 2019 Aug 2;39(8):BSR20190377. doi: 10.1042/BSR20190377. Print 2019 Aug 30. Biosci Rep. 2019. PMID: 31341010 Free PMC article. Review.
-
How Activin A Became a Therapeutic Target in Fibrodysplasia Ossificans Progressiva.Biomolecules. 2024 Jan 12;14(1):101. doi: 10.3390/biom14010101. Biomolecules. 2024. PMID: 38254701 Free PMC article. Review.
Cited by
-
Functional Testing of Bone Morphogenetic Protein (BMP) Pathway Variants Identified on Whole-Exome Sequencing in a Patient with Delayed-Onset Fibrodysplasia Ossificans Progressiva (FOP) Using ACVR1R206H -Specific Human Cellular and Zebrafish Models.J Bone Miner Res. 2022 Nov;37(11):2058-2076. doi: 10.1002/jbmr.4711. Epub 2022 Nov 15. J Bone Miner Res. 2022. PMID: 36153796 Free PMC article.
-
AAV-Mediated Targeting of the Activin A-ACVR1R206H Signaling in Fibrodysplasia Ossificans Progressiva.Biomolecules. 2023 Sep 8;13(9):1364. doi: 10.3390/biom13091364. Biomolecules. 2023. PMID: 37759764 Free PMC article.
-
Pathogenic ACVR1R206H activation by Activin A-induced receptor clustering and autophosphorylation.EMBO J. 2021 Jul 15;40(14):e106317. doi: 10.15252/embj.2020106317. Epub 2021 May 18. EMBO J. 2021. PMID: 34003511 Free PMC article.
-
Garetosmab in fibrodysplasia ossificans progressiva: a randomized, double-blind, placebo-controlled phase 2 trial.Nat Med. 2023 Oct;29(10):2615-2624. doi: 10.1038/s41591-023-02561-8. Epub 2023 Sep 28. Nat Med. 2023. PMID: 37770652 Free PMC article. Clinical Trial.
-
Cell Senescence in Heterotopic Ossification.Biomolecules. 2024 Apr 16;14(4):485. doi: 10.3390/biom14040485. Biomolecules. 2024. PMID: 38672501 Free PMC article. Review.
References
-
- Hatsell SJ, Idone V, Wolken DM, Huang L, Kim HJ, Wang L, Wen X, Nannuru KC, Jimenez J, Xie L, Das N, Makhoul G, Chernomorsky R, D’Ambrosio D, Corpina RA, Schoenherr CJ, Feeley K, Yu PB, Yancopoulos GD, Murphy AJ, Economides AN, ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A, Sci. Transl. Med 7 (303) (2015) (303ra137). - PMC - PubMed
-
- Dey D, Bagarova J, Hatsell SJ, Armstrong KA, Huang L, Ermann J, Vonner AJ, Shen Y, Mohedas AH, Lee A, Eekhoff EMW, van Schie A, Demay MB, Keller C, Wagers AJ, Economides AN, Yu PB, Two tissue-resident progenitor lineages drive distinct phenotypes of heterotopic ossification, Sci. Transl. Med 8 (366) (2016) (366ra163–366ra163). - PMC - PubMed
-
- Kaplan FS, Glaser DL, Shore EM, Deirmengian GK, Gupta R, Delai P, Morhart R, Smith R, Le Merrer M, Rogers JG, Connor JM, Kitterman JA, The phenotype of fibrodysplasia ossificans progressiva, Clin. Rev. Bone Miner. Metab 3 (3) (2005) 183–188.
-
- Kaplan FS, Groppe JC, Seemann P, Pignolo RJ, Shore EM, Fibrodysplasia ossificans progressiva: developmental implications of a novel metamorphogene, in: Bronner F, Farach-Carson MC, Roach HI (Eds.), Bone and Development, Springer London, London: 2010, pp. 233–249.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
