

Clinical course of sly syndrome (mucopolysaccharidosis type VII)
- PMID: 26908836
- PMCID: PMC4893087
- DOI: 10.1136/jmedgenet-2015-103322
Clinical course of sly syndrome (mucopolysaccharidosis type VII)
Abstract
Background: Mucopolysaccharidosis VII (MPS VII) is an ultra-rare disease characterised by the deficiency of β-glucuronidase (GUS). Patients' phenotypes vary from severe forms with hydrops fetalis, skeletal dysplasia and mental retardation to milder forms with fewer manifestations and mild skeletal abnormalities. Accurate assessments on the frequency and clinical characteristics of the disease have been scarce. The aim of this study was to collect such data.
Methods: We have conducted a survey of physicians to document the medical history of patients with MPS VII. The survey included anonymous information on patient demographics, family history, mode of diagnosis, age of onset, signs and symptoms, severity, management, clinical features and natural progression of the disease.
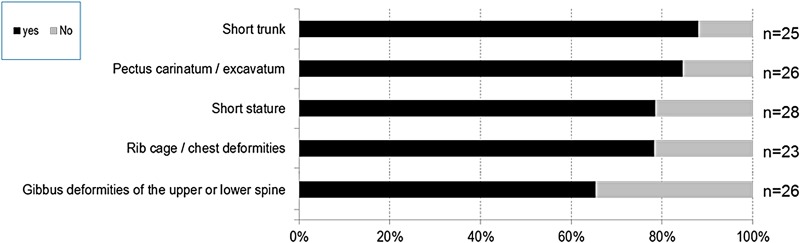
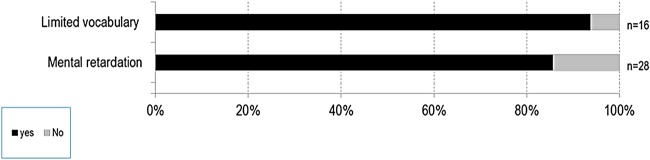
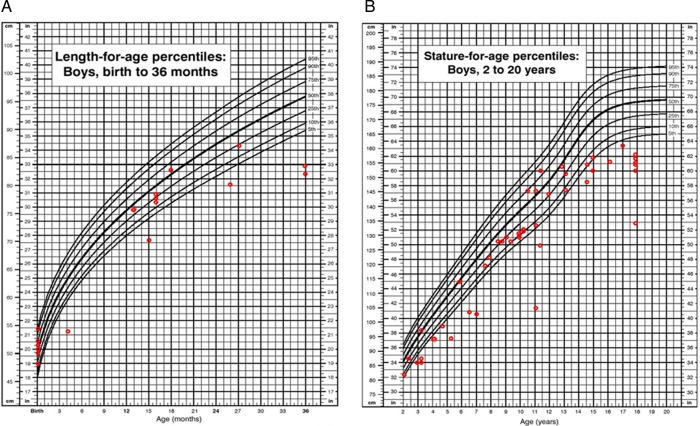
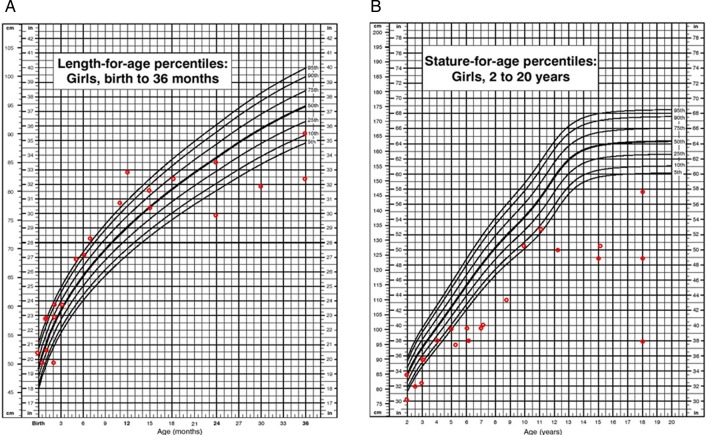
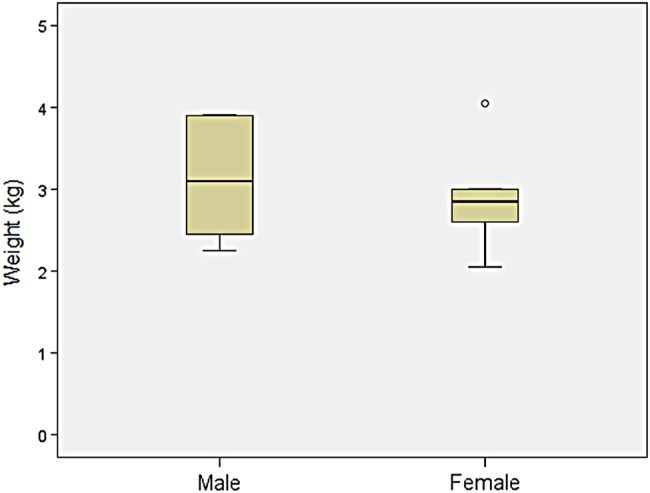
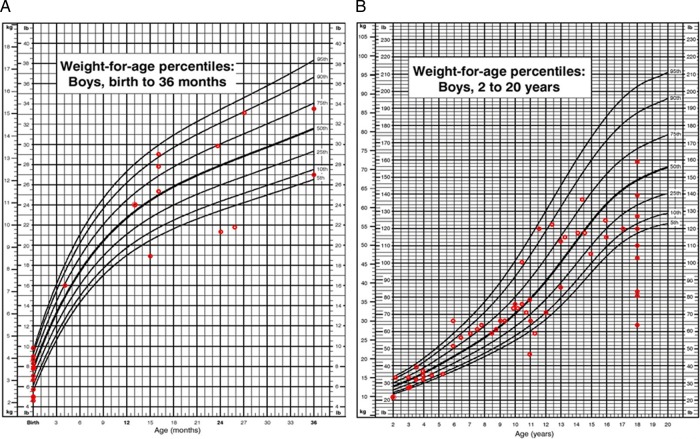
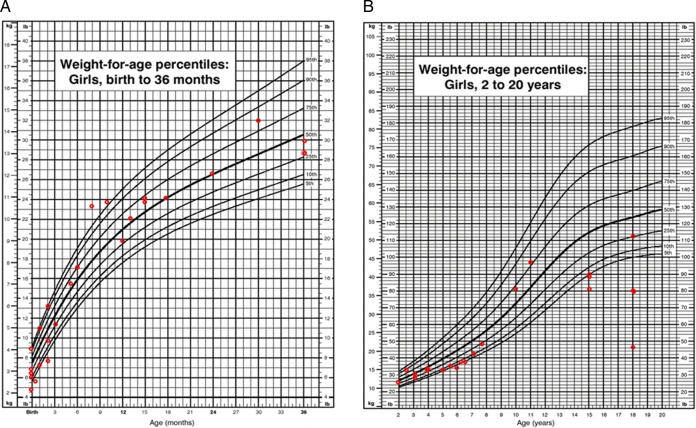
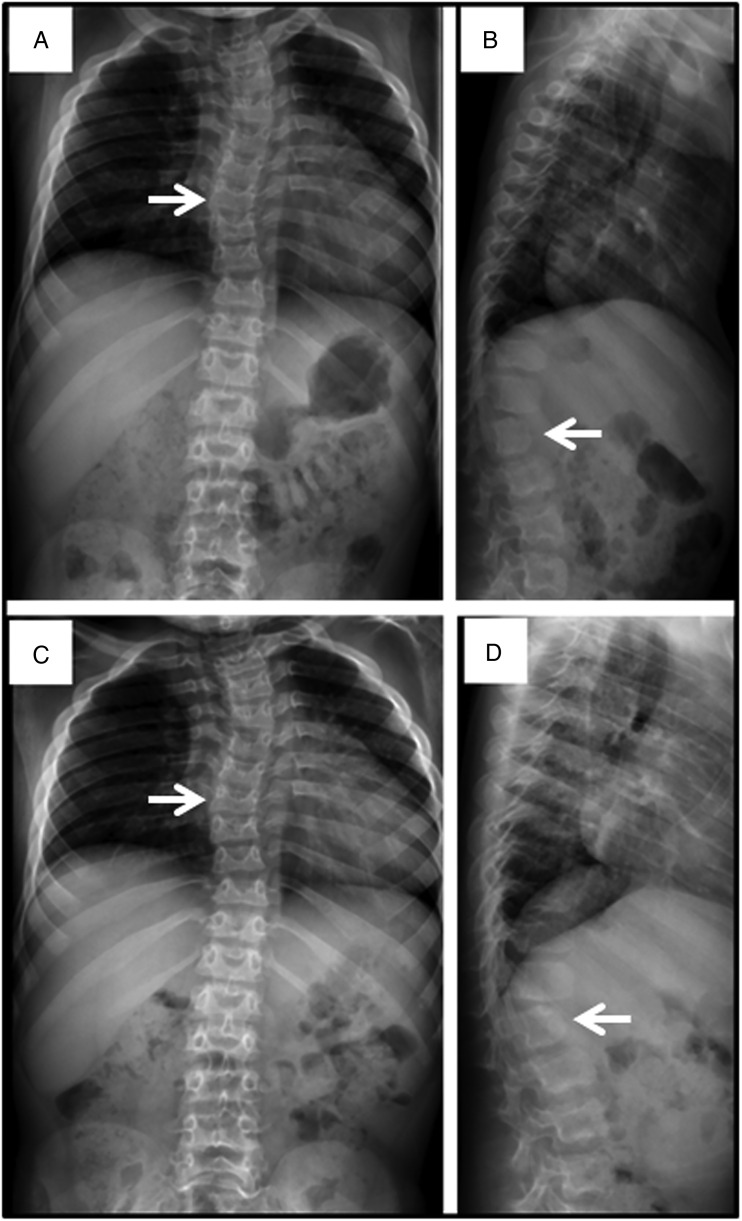
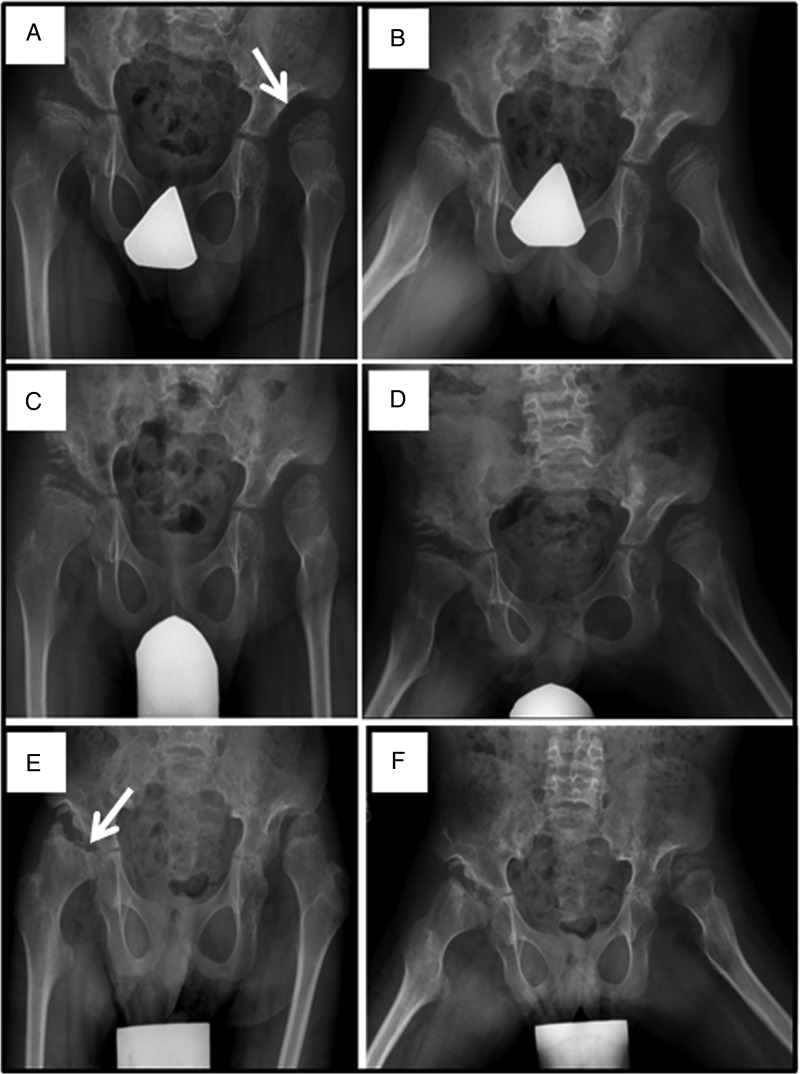
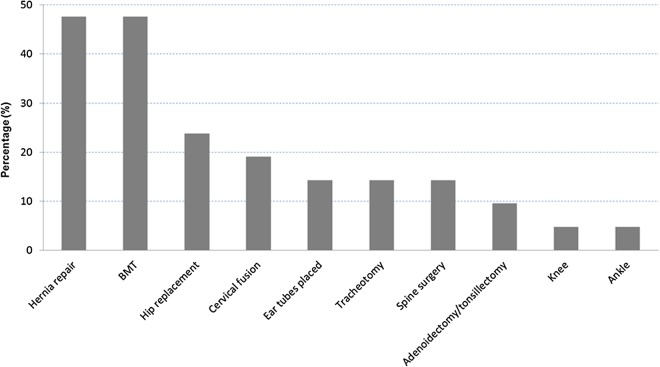
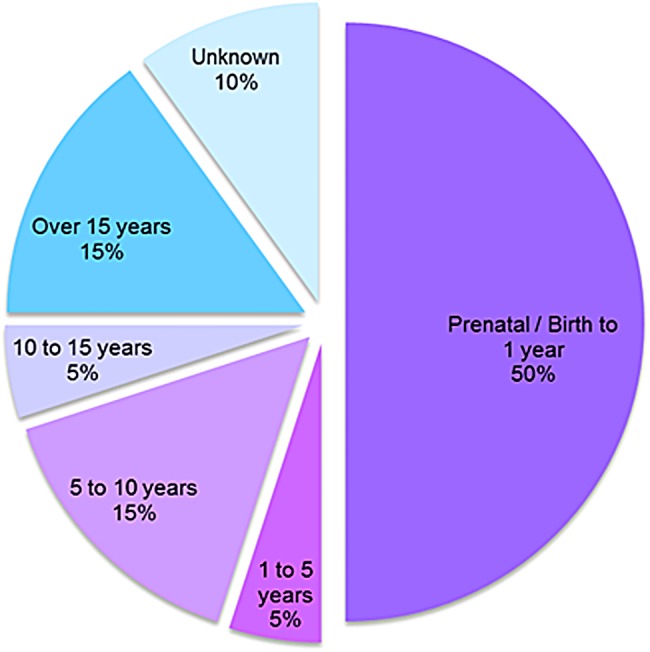
Results: We collected information on 56 patients from 11 countries. Patients with MPS VII were classified based on their phenotype into three different groups: (1) neonatal non-immune hydrops fetalis (NIHF) (n=10), (2) Infantile or adolescent form with history of hydrops fetalis (n=13) and (3) Infantile or adolescent form without known hydrops fetalis (n=33). Thirteen patients with MPS VII who had the infantile form with history of hydrops fetalis and survived childhood, had a wide range of clinical manifestations from mild to severe. Five patients underwent bone marrow transplantation and one patient underwent enzyme replacement therapy with recombinant human GUS.
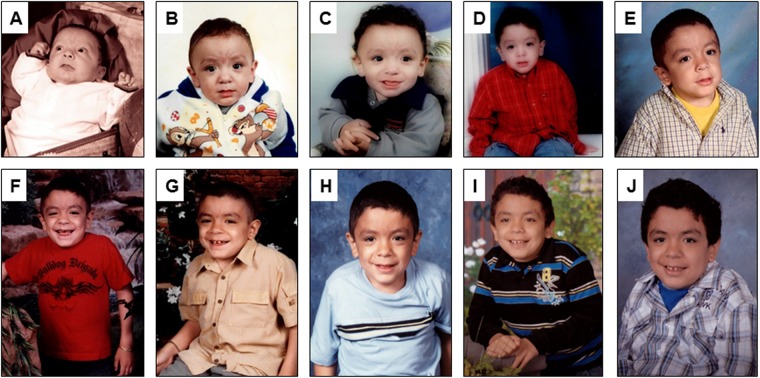
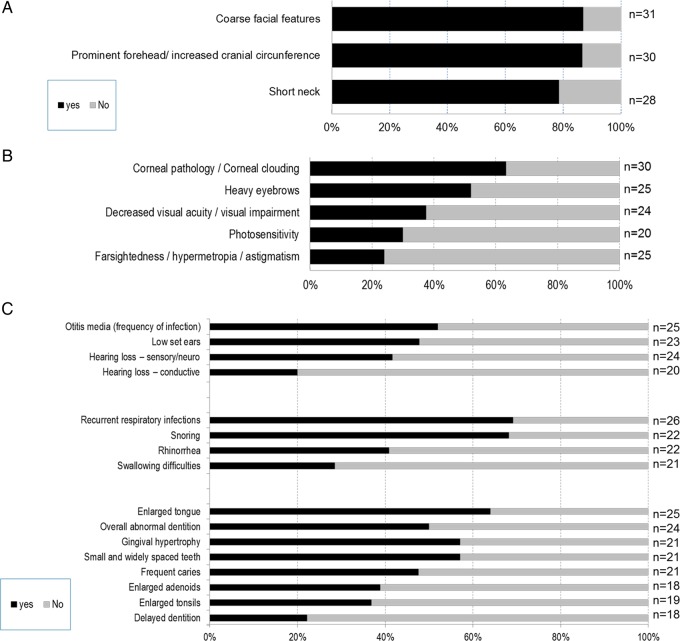
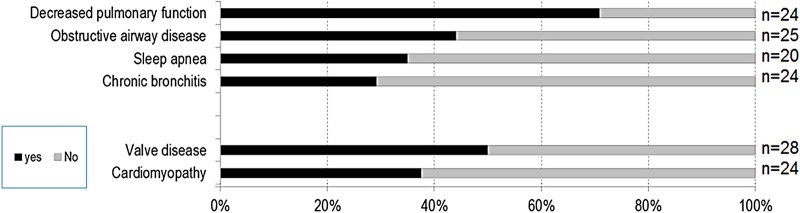
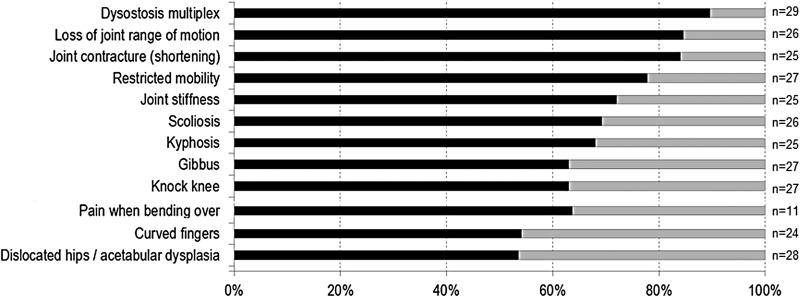
Conclusions: MPS VII is a pan-ethnic inherited lysosomal storage disease with considerable phenotypical heterogeneity. Most patients have short stature, skeletal dysplasia, hepatosplenomegaly, hernias, cardiac involvement, pulmonary insufficiency and cognitive impairment. In these respects it resembles MPS I and MPS II. In MPS VII, however, one unique and distinguishing clinical feature is the unexpectedly high proportion of patients (41%) that had a history of NIHF. Presence of NIHF does not, by itself, predict the eventual severity of the clinical course, if the patient survives infancy.
Keywords: Clinical genetics; Genetics; Metabolic disorders.
Published by the BMJ Publishing Group Limited. For permission to use (where not already granted under a licence) please go to http://www.bmj.com/company/products-services/rights-and-licensing/
Figures
Similar articles
-
Mucopolysaccharidosis type VII (Sly syndrome) - What do we know?Mol Genet Metab. 2024 Mar;141(3):108145. doi: 10.1016/j.ymgme.2024.108145. Epub 2024 Jan 17. Mol Genet Metab. 2024. PMID: 38301529 Review.
-
Disease characteristics, effectiveness, and safety of vestronidase alfa for the treatment of patients with mucopolysaccharidosis VII in a novel, longitudinal, multicenter disease monitoring program.Orphanet J Rare Dis. 2024 May 7;19(1):189. doi: 10.1186/s13023-024-03176-z. Orphanet J Rare Dis. 2024. PMID: 38715031 Free PMC article.
-
Pathway to diagnosis and burden of illness in mucopolysaccharidosis type VII - a European caregiver survey.Orphanet J Rare Dis. 2019 Nov 14;14(1):254. doi: 10.1186/s13023-019-1233-z. Orphanet J Rare Dis. 2019. PMID: 31727109 Free PMC article.
-
A new case report of severe mucopolysaccharidosis type VII: diagnosis, treatment with haematopoietic cell transplantation and prenatal diagnosis in a second pregnancy.Ital J Pediatr. 2018 Nov 16;44(Suppl 2):128. doi: 10.1186/s13052-018-0566-x. Ital J Pediatr. 2018. PMID: 30442200 Free PMC article.
-
Mucopolysaccharidosis VII in Brazil: natural history and clinical findings.Orphanet J Rare Dis. 2021 May 22;16(1):238. doi: 10.1186/s13023-021-01870-w. Orphanet J Rare Dis. 2021. PMID: 34022924 Free PMC article. Review.
Cited by
-
Intravenous Enzyme Replacement Therapy in Mucopolysaccharidoses: Clinical Effectiveness and Limitations.Int J Mol Sci. 2020 Apr 23;21(8):2975. doi: 10.3390/ijms21082975. Int J Mol Sci. 2020. PMID: 32340185 Free PMC article. Review.
-
Elevation of glycosaminoglycans in the amniotic fluid of a fetus with mucopolysaccharidosis VII.Prenat Diagn. 2017 May;37(5):435-439. doi: 10.1002/pd.5028. Epub 2017 Mar 12. Prenat Diagn. 2017. PMID: 28207930 Free PMC article.
-
Dental-craniofacial manifestation and treatment of rare diseases.Int J Oral Sci. 2019 Feb 20;11(1):9. doi: 10.1038/s41368-018-0041-y. Int J Oral Sci. 2019. PMID: 30783081 Free PMC article. Review.
-
Hearing Loss in Mucopolysaccharidoses: Current Knowledge and Future Directions.Diagnostics (Basel). 2020 Aug 4;10(8):554. doi: 10.3390/diagnostics10080554. Diagnostics (Basel). 2020. PMID: 32759694 Free PMC article. Review.
-
Lysosomal storage disease overview.Ann Transl Med. 2018 Dec;6(24):476. doi: 10.21037/atm.2018.11.39. Ann Transl Med. 2018. PMID: 30740407 Free PMC article. Review.
References
-
- Neufeld EF, Muenzer J. The Mucopolysaccharidoses. In: Scriver CR, Beaudet A, Sly WS, et al.. eds The metabolic and molecular basis of inherited disease. Vol III McGraw-Hill, 2001:3421–52.
-
- Guibaud P, Maire I, Goddon R, Teyssier G, Zabot MT, Mandon G. [Mucopolysaccharidesis Type VII resulting from beta-glucuronidase deficiency. Report of one family]. J Genet Hum 1979;27:29–43. - PubMed