

Congenital disorders of autophagy: an emerging novel class of inborn errors of neuro-metabolism
- PMID: 26715604
- PMCID: PMC5841365
- DOI: 10.1093/brain/awv371
Congenital disorders of autophagy: an emerging novel class of inborn errors of neuro-metabolism
Abstract
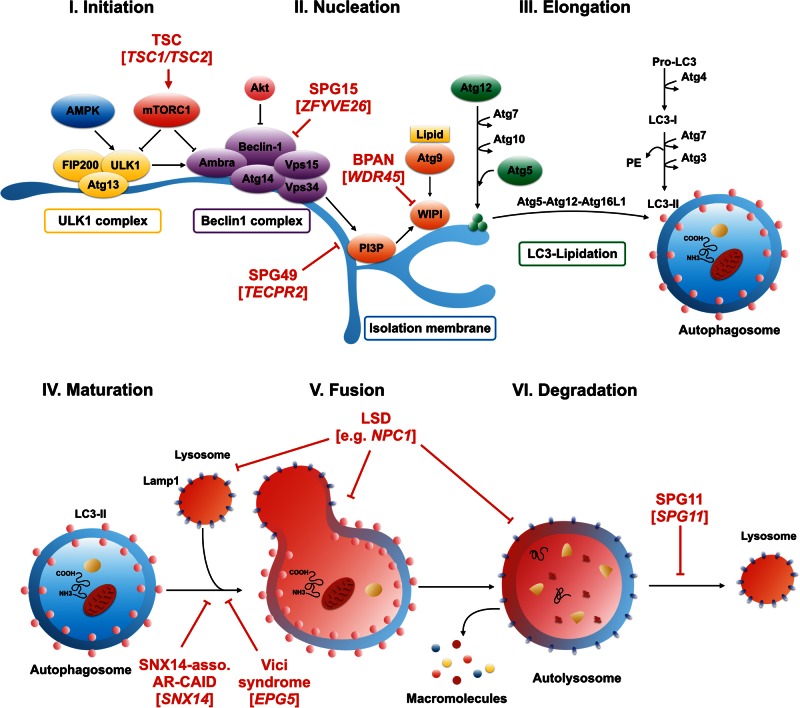
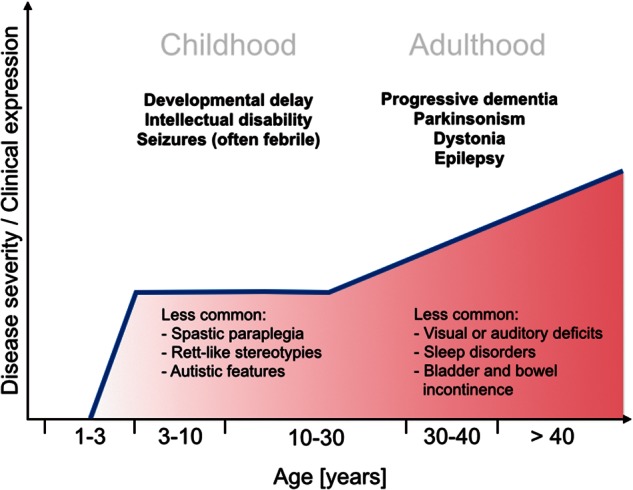
Single gene disorders of the autophagy pathway are an emerging, novel and diverse group of multisystem diseases in children. Clinically, these disorders prominently affect the central nervous system at various stages of development, leading to brain malformations, developmental delay, intellectual disability, epilepsy, movement disorders, and neurodegeneration, among others. Frequent early and severe involvement of the central nervous system puts the paediatric neurologist, neurogeneticist, and neurometabolic specialist at the forefront of recognizing and treating these rare conditions. On a molecular level, mutations in key autophagy genes map to different stages of this highly conserved pathway and thus lead to impairment in isolation membrane (or phagophore) and autophagosome formation, maturation, or autophagosome-lysosome fusion. Here we discuss 'congenital disorders of autophagy' as an emerging subclass of inborn errors of metabolism by using the examples of six recently identified monogenic diseases: EPG5-related Vici syndrome, beta-propeller protein-associated neurodegeneration due to mutations in WDR45, SNX14-associated autosomal-recessive cerebellar ataxia and intellectual disability syndrome, and three forms of hereditary spastic paraplegia, SPG11, SPG15 and SPG49 caused by SPG11, ZFYVE26 and TECPR2 mutations, respectively. We also highlight associations between defective autophagy and other inborn errors of metabolism such as lysosomal storage diseases and neurodevelopmental diseases associated with the mTOR pathway, which may be included in the wider spectrum of autophagy-related diseases from a pathobiological point of view. By exploring these emerging themes in disease pathogenesis and underlying pathophysiological mechanisms, we discuss how congenital disorders of autophagy inform our understanding of the importance of this fascinating cellular pathway for central nervous system biology and disease. Finally, we review the concept of modulating autophagy as a therapeutic target and argue that congenital disorders of autophagy provide a unique genetic perspective on the possibilities and challenges of pathway-specific drug development.
Keywords: autophagy; inborn errors of metabolism; mammalian target of rapamycin (mTOR); neurodegeneration; neurodevelopment.
© The Author (2015). Published by Oxford University Press on behalf of the Guarantors of Brain. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
Similar articles
-
Novel insights into the clinical and molecular spectrum of congenital disorders of autophagy.J Inherit Metab Dis. 2020 Jan;43(1):51-62. doi: 10.1002/jimd.12084. Epub 2019 Apr 8. J Inherit Metab Dis. 2020. PMID: 30854657 Review.
-
Congenital Disorders of Autophagy: What a Pediatric Neurologist Should Know.Neuropediatrics. 2018 Feb;49(1):18-25. doi: 10.1055/s-0037-1608652. Epub 2017 Nov 7. Neuropediatrics. 2018. PMID: 29112993 Review.
-
ZFYVE26/SPASTIZIN and SPG11/SPATACSIN mutations in hereditary spastic paraplegia types AR-SPG15 and AR-SPG11 have different effects on autophagy and endocytosis.Autophagy. 2019 Jan;15(1):34-57. doi: 10.1080/15548627.2018.1507438. Epub 2018 Sep 13. Autophagy. 2019. PMID: 30081747 Free PMC article.
-
The spectrum of neurodevelopmental, neuromuscular and neurodegenerative disorders due to defective autophagy.Autophagy. 2022 Mar;18(3):496-517. doi: 10.1080/15548627.2021.1943177. Epub 2021 Aug 19. Autophagy. 2022. PMID: 34130600 Free PMC article.
-
EPG5-related Vici syndrome: a paradigm of neurodevelopmental disorders with defective autophagy.Brain. 2016 Mar;139(Pt 3):765-81. doi: 10.1093/brain/awv393. Brain. 2016. PMID: 26917586 Free PMC article.
Cited by
-
Molecular Basis of Neuronal Autophagy in Ageing: Insights from Caenorhabditis elegans.Cells. 2021 Mar 21;10(3):694. doi: 10.3390/cells10030694. Cells. 2021. PMID: 33800981 Free PMC article. Review.
-
High-content screening identifies a small molecule that restores AP-4-dependent protein trafficking in neuronal models of AP-4-associated hereditary spastic paraplegia.Nat Commun. 2024 Jan 17;15(1):584. doi: 10.1038/s41467-023-44264-1. Nat Commun. 2024. PMID: 38233389 Free PMC article.
-
OSBPL2 mutations impair autophagy and lead to hearing loss, potentially remedied by rapamycin.Autophagy. 2022 Nov;18(11):2593-2614. doi: 10.1080/15548627.2022.2040891. Epub 2022 Mar 6. Autophagy. 2022. PMID: 35253614 Free PMC article.
-
mTOR and autophagy pathways are dysregulated in murine and human models of Schaaf-Yang syndrome.Sci Rep. 2019 Nov 4;9(1):15935. doi: 10.1038/s41598-019-52287-2. Sci Rep. 2019. PMID: 31685878 Free PMC article.
-
Eye involvement in inherited metabolic disorders.Ther Adv Ophthalmol. 2020 Dec 29;12:2515841420979109. doi: 10.1177/2515841420979109. eCollection 2020 Jan-Dec. Ther Adv Ophthalmol. 2020. PMID: 33447730 Free PMC article. Review.
References
-
- Al-Owain M, Al-Hashem A, Al-Muhaizea M, Humaidan H, Al-Hindi H, Al-Homoud I, et al. Vici syndrome associated with unilateral lung hypoplasia and myopathy. Am J Med Genet A 2010; 152A: 1849–53. - PubMed
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
