

Mutation screen reveals novel variants and expands the phenotypes associated with DYNC1H1
- PMID: 26100331
- PMCID: PMC4573829
- DOI: 10.1007/s00415-015-7727-2
Mutation screen reveals novel variants and expands the phenotypes associated with DYNC1H1
Abstract
Dynein, cytoplasmic 1, heavy chain 1 (DYNC1H1) encodes a necessary subunit of the cytoplasmic dynein complex, which traffics cargo along microtubules. Dominant DYNC1H1 mutations are implicated in neural diseases, including spinal muscular atrophy with lower extremity dominance (SMA-LED), intellectual disability with neuronal migration defects, malformations of cortical development, and Charcot-Marie-Tooth disease, type 2O. We hypothesized that additional variants could be found in these and novel motoneuron and related diseases. Therefore, we analyzed our database of 1024 whole exome sequencing samples of motoneuron and related diseases for novel single nucleotide variations. We filtered these results for significant variants, which were further screened using segregation analysis in available family members. Analysis revealed six novel, rare, and highly conserved variants. Three of these are likely pathogenic and encompass a broad phenotypic spectrum with distinct disease clusters. Our findings suggest that DYNC1H1 variants can cause not only lower, but also upper motor neuron disease. It thus adds DYNC1H1 to the growing list of spastic paraplegia related genes in microtubule-dependent motor protein pathways.
Keywords: DYNC1H1; Epilepsy; Gastric volvulus; Peripheral neuropathy; Spastic paraplegia; Spinal muscular atrophy.
Conflict of interest statement
These authors declare that they have no conflict of interest.
Figures
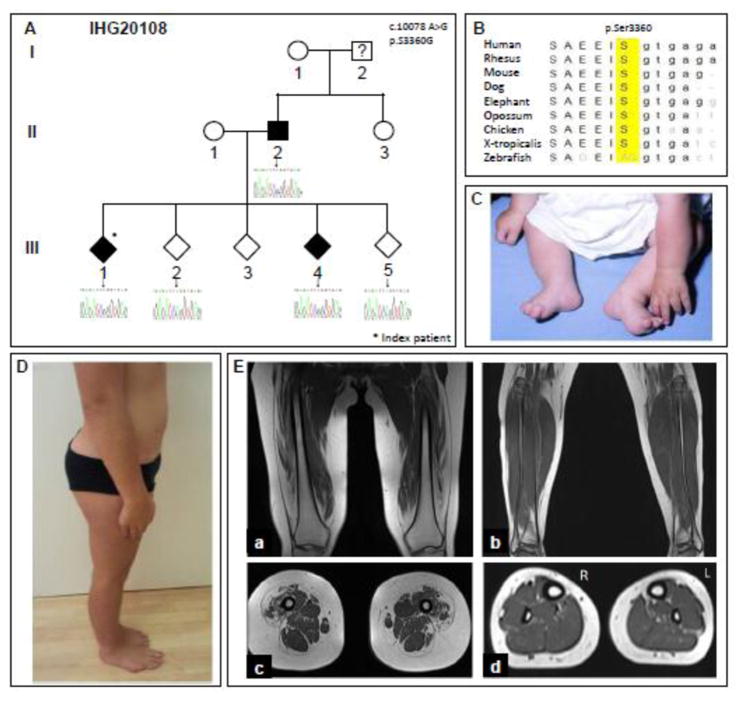
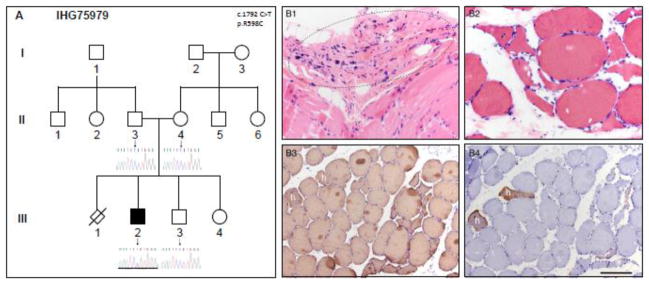
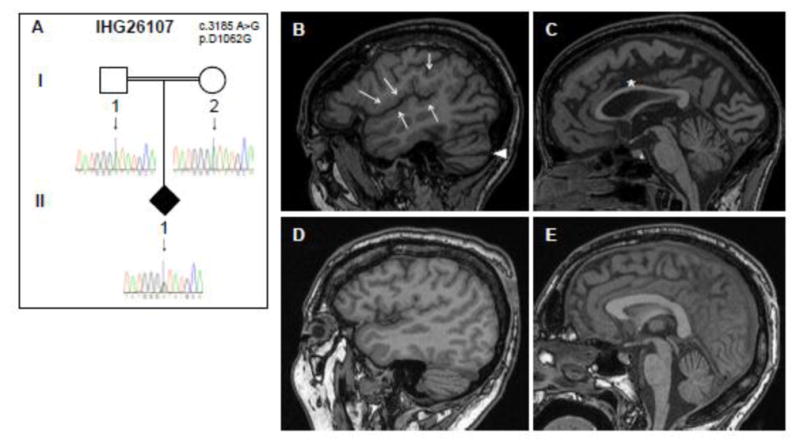
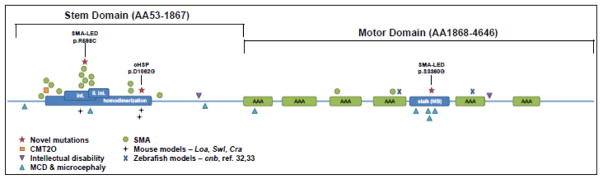
Similar articles
-
Novel mutations in the DYNC1H1 tail domain refine the genetic and clinical spectrum of dyneinopathies.Hum Mutat. 2015 Mar;36(3):287-91. doi: 10.1002/humu.22744. Hum Mutat. 2015. PMID: 25512093
-
Novel dynein DYNC1H1 neck and motor domain mutations link distal spinal muscular atrophy and abnormal cortical development.Hum Mutat. 2014 Mar;35(3):298-302. doi: 10.1002/humu.22491. Epub 2014 Jan 3. Hum Mutat. 2014. PMID: 24307404 Free PMC article.
-
Mutations in the tail domain of DYNC1H1 cause dominant spinal muscular atrophy.Neurology. 2012 May 29;78(22):1714-20. doi: 10.1212/WNL.0b013e3182556c05. Epub 2012 Mar 28. Neurology. 2012. PMID: 22459677 Free PMC article.
-
Whole-exome sequencing identifies a novel de novo variant in DYNC1H in a patient with intractable epilepsy.Neurol Sci. 2022 Apr;43(4):2853-2858. doi: 10.1007/s10072-021-05824-9. Epub 2022 Jan 28. Neurol Sci. 2022. PMID: 35088241 Review.
-
VRK1 variants at the cross road of Cajal body neuropathogenic mechanisms in distal neuropathies and motor neuron diseases.Neurobiol Dis. 2023 Jul;183:106172. doi: 10.1016/j.nbd.2023.106172. Epub 2023 May 29. Neurobiol Dis. 2023. PMID: 37257665 Review.
Cited by
-
Update on recent advances in amyotrophic lateral sclerosis.J Neurol. 2024 Jul;271(7):4693-4723. doi: 10.1007/s00415-024-12435-9. Epub 2024 May 27. J Neurol. 2024. PMID: 38802624 Free PMC article. Review.
-
The clinical-phenotype continuum in DYNC1H1-related disorders-genomic profiling and proposal for a novel classification.J Hum Genet. 2020 Nov;65(11):1003-1017. doi: 10.1038/s10038-020-0803-1. Epub 2020 Aug 12. J Hum Genet. 2020. PMID: 32788638 Free PMC article.
-
Cryo-EM Reveals How Human Cytoplasmic Dynein Is Auto-inhibited and Activated.Cell. 2017 Jun 15;169(7):1303-1314.e18. doi: 10.1016/j.cell.2017.05.025. Epub 2017 Jun 8. Cell. 2017. PMID: 28602352 Free PMC article.
-
The regulatory function of the AAA4 ATPase domain of cytoplasmic dynein.Nat Commun. 2020 Nov 23;11(1):5952. doi: 10.1038/s41467-020-19477-3. Nat Commun. 2020. PMID: 33230227 Free PMC article.
-
Molecular mechanism of cytoplasmic dynein tension sensing.Nat Commun. 2019 Jul 26;10(1):3332. doi: 10.1038/s41467-019-11231-8. Nat Commun. 2019. PMID: 31350388 Free PMC article.
References
-
- Vissers LELM, de Ligt J, Gilissen C, et al. A de novo paradigm for mental retardation. Nat Genet. 2010;42 (12):1109–1112. - PubMed
-
- Willemsen MH, Vissers LEL, Willemsen MAAP, et al. Mutations in DYNC1H1 cause severe intellectual disability with neuronal migration defects. J Med Genet. 2012;49 (3):179–183. - PubMed
-
- Tsurusaki Y, Saitoh S, Tomizawa K, et al. A DYNC1H1 mutation causes a dominant spinal muscular atrophy with lower extremity predominance. Neurogenetics. 2012;13 (4):327–332. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U54NS053672/NS/NINDS NIH HHS/United States
- U54 NS065712/NS/NINDS NIH HHS/United States
- K08 NS075094/NS/NINDS NIH HHS/United States
- U54NS0657/NS/NINDS NIH HHS/United States
- P 23223/FWF_/Austrian Science Fund FWF/Austria
- K08-NS075094/NS/NINDS NIH HHS/United States
- R01NS075764/NS/NINDS NIH HHS/United States
- U54 NS053672/NS/NINDS NIH HHS/United States
- R01NS072248/NS/NINDS NIH HHS/United States
- R01 NS072248/NS/NINDS NIH HHS/United States
- R01 NS075764/NS/NINDS NIH HHS/United States
- P 27634/FWF_/Austrian Science Fund FWF/Austria
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
