

Genetic defects in dolichol metabolism
- PMID: 25270028
- PMCID: PMC4281381
- DOI: 10.1007/s10545-014-9760-1
Genetic defects in dolichol metabolism
Abstract
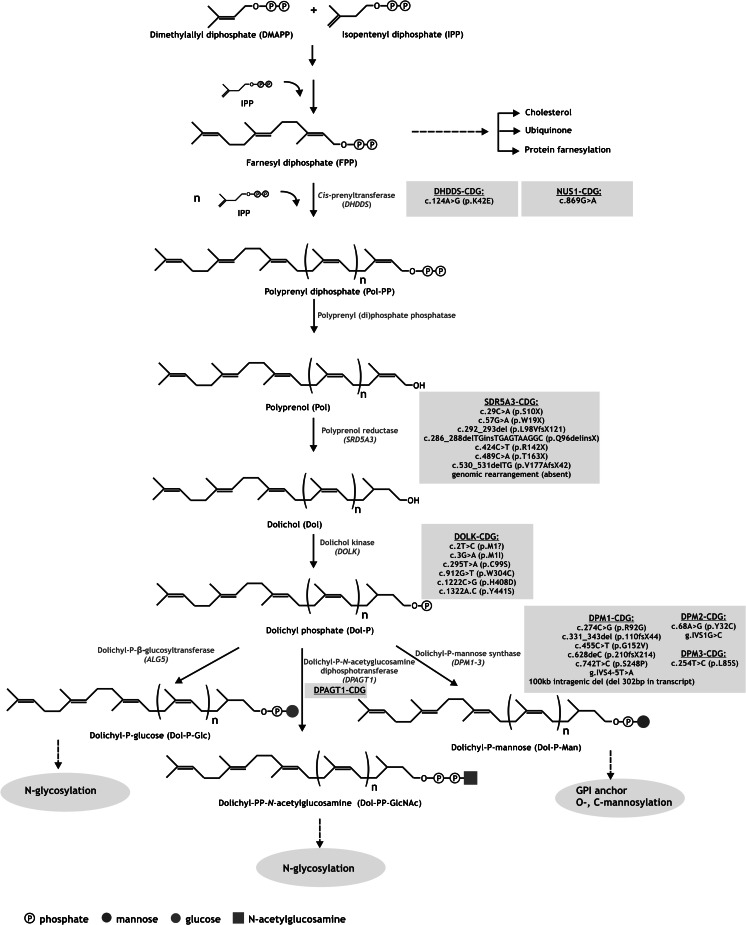
Congenital disorders of glycosylation (CDG) comprise a group of inborn errors of metabolism with abnormal glycosylation of proteins and lipids. Patients with defective protein N-glycosylation are identified in routine metabolic screening via analysis of serum transferrin glycosylation. Defects in the assembly of the dolichol linked Glc(3)Man(9)GlcNAc(2) glycan and its transfer to proteins lead to the (partial) absence of complete glycans on proteins. These defects are called CDG-I and are located in the endoplasmic reticulum (ER) or cytoplasm. Defects in the subsequent processing of protein bound glycans result in the presence of truncated glycans on proteins. These defects are called CDG-II and the enzymes involved are located mainly in the Golgi apparatus. In recent years, human defects have been identified in dolichol biosynthesis genes within the group of CDG-I patients. This has increased interest in dolichol metabolism, has resulted in specific recognizable clinical symptoms in CDG-I and has offered new mechanistic insights in dolichol biosynthesis. We here review its biosynthetic pathways, the clinical and biochemical phenotypes in dolichol-related CDG defects, up to the formation of dolichyl-P-mannose (Dol-P-Man), and discuss existing evidence of regulatory networks in dolichol metabolism to provide an outlook on therapeutic strategies.
Figures
Similar articles
-
Congenital Disorders of N-Linked Glycosylation and Multiple Pathway Overview.2005 Aug 15 [updated 2017 Jan 12]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. 2005 Aug 15 [updated 2017 Jan 12]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 20301507 Free Books & Documents. Review.
-
Improvement of dolichol-linked oligosaccharide biosynthesis by the squalene synthase inhibitor zaragozic acid.J Biol Chem. 2011 Feb 25;286(8):6085-91. doi: 10.1074/jbc.M110.165795. Epub 2010 Dec 23. J Biol Chem. 2011. PMID: 21183681 Free PMC article.
-
Regulation of dolichol-linked glycosylation.Glycoconj J. 2013 Jan;30(1):51-6. doi: 10.1007/s10719-012-9417-y. Epub 2012 Jun 21. Glycoconj J. 2013. PMID: 22717794 Review.
-
A new role for dolichol isoform profile in the diagnostics of CDG disorders.Clin Chim Acta. 2020 Aug;507:88-93. doi: 10.1016/j.cca.2020.04.012. Epub 2020 Apr 11. Clin Chim Acta. 2020. PMID: 32289257
-
Toward understanding tissue-specific symptoms in dolichol-phosphate-mannose synthesis disorders; insight from DPM3-CDG.J Inherit Metab Dis. 2019 Sep;42(5):984-992. doi: 10.1002/jimd.12095. Epub 2019 Apr 23. J Inherit Metab Dis. 2019. PMID: 30931530
Cited by
-
Aptamer-Based Imaging of Polyisoprenoids in the Malaria Parasite.Molecules. 2023 Dec 28;29(1):178. doi: 10.3390/molecules29010178. Molecules. 2023. PMID: 38202761 Free PMC article.
-
SRD5A3-CDG: Emerging Phenotypic Features of an Ultrarare CDG Subtype.Front Genet. 2021 Dec 1;12:737094. doi: 10.3389/fgene.2021.737094. eCollection 2021. Front Genet. 2021. PMID: 34925443 Free PMC article.
-
Dolichol kinases from yeast, nematode and human can replace each other and exchange their domains creating active chimeric enzymes in yeast.PLoS One. 2024 Nov 7;19(11):e0313330. doi: 10.1371/journal.pone.0313330. eCollection 2024. PLoS One. 2024. PMID: 39509371 Free PMC article.
-
Identification and characterization of transcriptional control region of the human beta 1,4-mannosyltransferase gene.Cytotechnology. 2017 Jun;69(3):417-434. doi: 10.1007/s10616-015-9929-y. Epub 2015 Nov 25. Cytotechnology. 2017. PMID: 26608959 Free PMC article.
-
Selective Ablation of Dehydrodolichyl Diphosphate Synthase in Murine Retinal Pigment Epithelium (RPE) Causes RPE Atrophy and Retinal Degeneration.Cells. 2020 Mar 21;9(3):771. doi: 10.3390/cells9030771. Cells. 2020. PMID: 32245241 Free PMC article.
References
-
- Adair WL, Jr, Cafmeyer N. Topography of dolichyl phosphate synthesis in rat liver microsomes. Transbilayer arrangement of dolichol kinase and long-chain prenyltransferase. Biochim Biophys Acta. 1983;751:21–26. - PubMed
-
- Adair WL, Jr, Cafmeyer N. Cell-cycle dependence of dolichyl phosphate biosynthesis. Arch Biochem Biophys. 1987;258(2):491–497. - PubMed
-
- Aebi M. N-linked protein glycosylation in the ER. Biochim Biophys Acta. 2013;1833(11):2430–2437. - PubMed
-
- Akhtar TA, Matsuba Y, Schauvinhold I, et al. The tomato cis-prenyltransferase gene family. Plant J. 2013;73(4):640–652. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
