

Variant non ketotic hyperglycinemia is caused by mutations in LIAS, BOLA3 and the novel gene GLRX5
- PMID: 24334290
- PMCID: PMC3914472
- DOI: 10.1093/brain/awt328
Variant non ketotic hyperglycinemia is caused by mutations in LIAS, BOLA3 and the novel gene GLRX5
Abstract
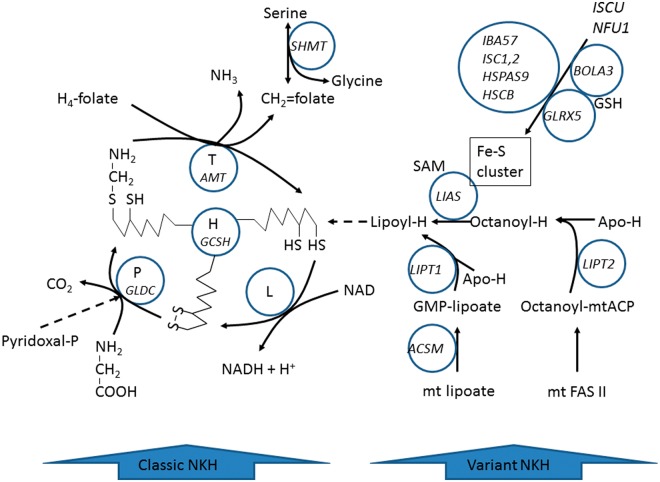
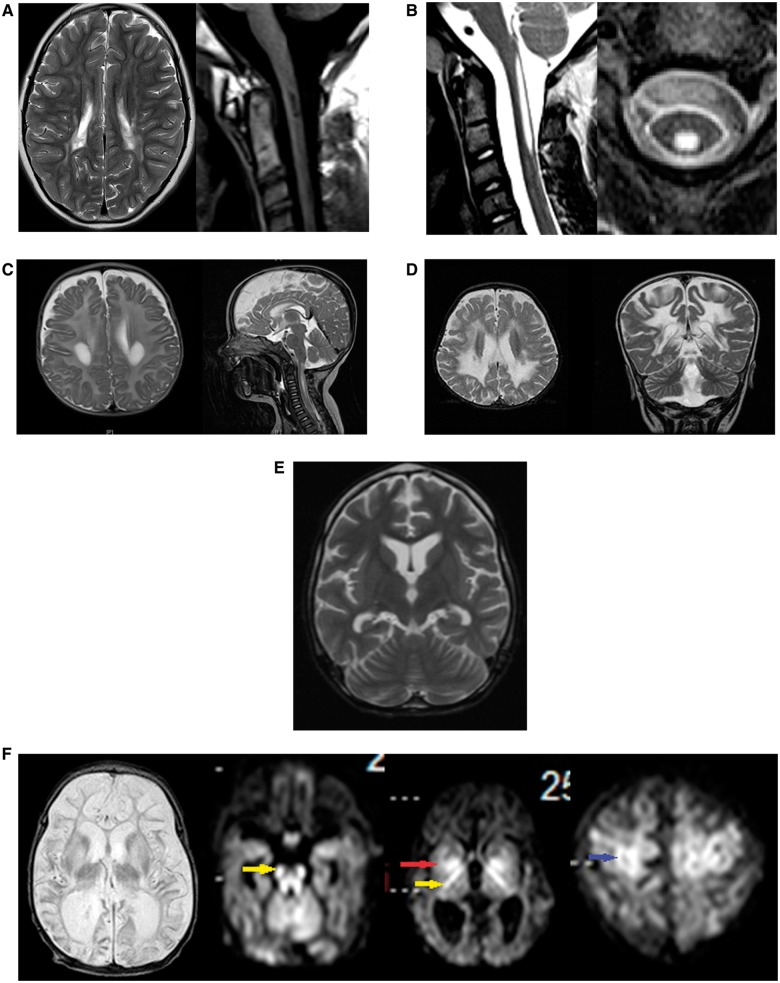
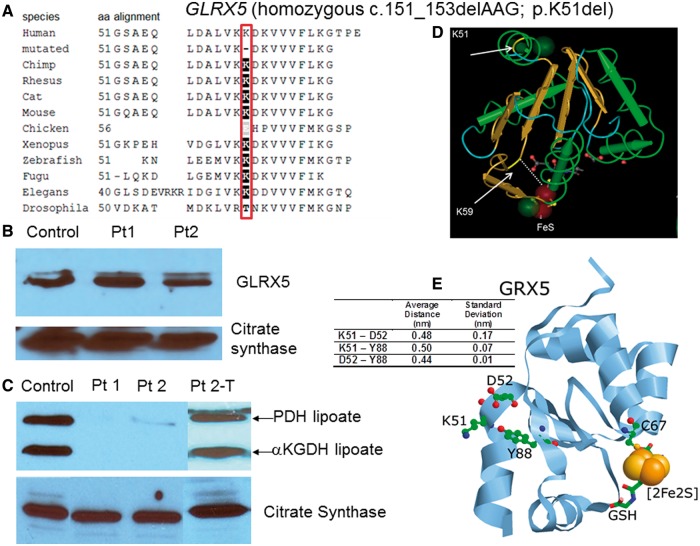
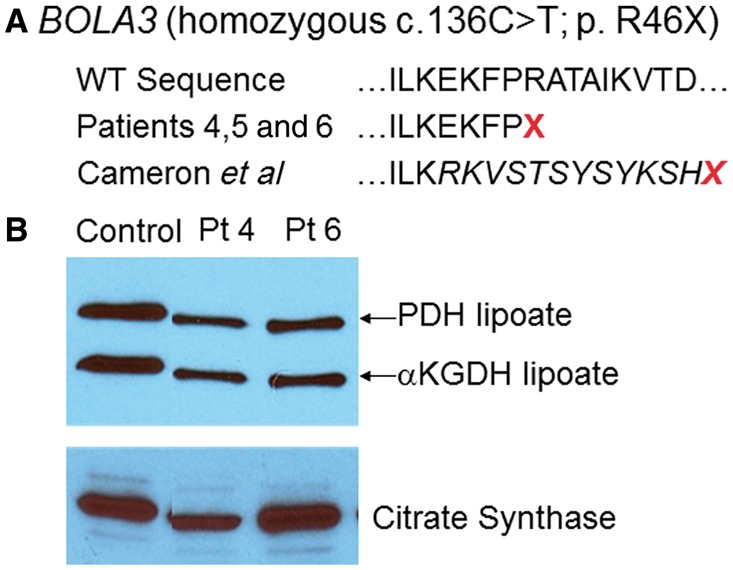
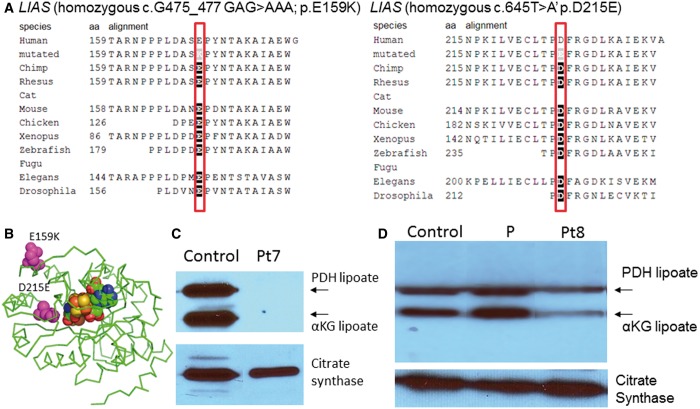
Patients with nonketotic hyperglycinemia and deficient glycine cleavage enzyme activity, but without mutations in AMT, GLDC or GCSH, the genes encoding its constituent proteins, constitute a clinical group which we call 'variant nonketotic hyperglycinemia'. We hypothesize that in some patients the aetiology involves genetic mutations that result in a deficiency of the cofactor lipoate, and sequenced genes involved in lipoate synthesis and iron-sulphur cluster biogenesis. Of 11 individuals identified with variant nonketotic hyperglycinemia, we were able to determine the genetic aetiology in eight patients and delineate the clinical and biochemical phenotypes. Mutations were identified in the genes for lipoate synthase (LIAS), BolA type 3 (BOLA3), and a novel gene glutaredoxin 5 (GLRX5). Patients with GLRX5-associated variant nonketotic hyperglycinemia had normal development with childhood-onset spastic paraplegia, spinal lesion, and optic atrophy. Clinical features of BOLA3-associated variant nonketotic hyperglycinemia include severe neurodegeneration after a period of normal development. Additional features include leukodystrophy, cardiomyopathy and optic atrophy. Patients with lipoate synthase-deficient variant nonketotic hyperglycinemia varied in severity from mild static encephalopathy to Leigh disease and cortical involvement. All patients had high serum and borderline elevated cerebrospinal fluid glycine and cerebrospinal fluid:plasma glycine ratio, and deficient glycine cleavage enzyme activity. They had low pyruvate dehydrogenase enzyme activity but most did not have lactic acidosis. Patients were deficient in lipoylation of mitochondrial proteins. There were minimal and inconsistent changes in cellular iron handling, and respiratory chain activity was unaffected. Identified mutations were phylogenetically conserved, and transfection with native genes corrected the biochemical deficiency proving pathogenicity. Treatments of cells with lipoate and with mitochondrially-targeted lipoate were unsuccessful at correcting the deficiency. The recognition of variant nonketotic hyperglycinemia is important for physicians evaluating patients with abnormalities in glycine as this will affect the genetic causation and genetic counselling, and provide prognostic information on the expected phenotypic course.
Keywords: iron-sulphur cluster; leukodystrophy; lipoic acid; nonketotic hyperglycinemia.
Figures
Similar articles
-
Case report: Unveiling genetic and phenotypic variability in Nonketotic hyperglycinemia: an atypical early onset case associated with a novel GLRX5 variant.Front Genet. 2024 Sep 11;15:1432272. doi: 10.3389/fgene.2024.1432272. eCollection 2024. Front Genet. 2024. PMID: 39323869 Free PMC article.
-
Lipoic acid biosynthesis defects.J Inherit Metab Dis. 2014 Jul;37(4):553-63. doi: 10.1007/s10545-014-9705-8. Epub 2014 Apr 29. J Inherit Metab Dis. 2014. PMID: 24777537 Review.
-
Nonketotic hyperglycinemia in captive-bred Vervet monkeys (Chlorocebus aethiops) with cataracts.J Med Primatol. 2019 Jun;48(3):161-165. doi: 10.1111/jmp.12400. Epub 2019 Feb 6. J Med Primatol. 2019. PMID: 30724368
-
Genotypic and phenotypic features in Turkish patients with classic nonketotic hyperglycinemia.Metab Brain Dis. 2021 Aug;36(6):1213-1222. doi: 10.1007/s11011-021-00718-3. Epub 2021 Apr 1. Metab Brain Dis. 2021. PMID: 33791923
-
Atypical variants of nonketotic hyperglycinemia.Mol Genet Metab. 2005 Sep-Oct;86(1-2):61-9. doi: 10.1016/j.ymgme.2005.07.016. Mol Genet Metab. 2005. PMID: 16157495 Review.
Cited by
-
Molecular Basis of Multiple Mitochondrial Dysfunctions Syndrome 2 Caused by CYS59TYR BOLA3 Mutation.Int J Mol Sci. 2021 May 3;22(9):4848. doi: 10.3390/ijms22094848. Int J Mol Sci. 2021. PMID: 34063696 Free PMC article.
-
Outlining the Complex Pathway of Mammalian Fe-S Cluster Biogenesis.Trends Biochem Sci. 2020 May;45(5):411-426. doi: 10.1016/j.tibs.2020.02.001. Epub 2020 Mar 6. Trends Biochem Sci. 2020. PMID: 32311335 Free PMC article. Review.
-
Mitochondrial lipoylation integrates age-associated decline in brown fat thermogenesis.Nat Metab. 2019 Sep;1(9):886-898. doi: 10.1038/s42255-019-0106-z. Epub 2019 Sep 16. Nat Metab. 2019. PMID: 32313871 Free PMC article.
-
Loss-of-function mutations in ISCA2 disrupt 4Fe-4S cluster machinery and cause a fatal leukodystrophy with hyperglycinemia and mtDNA depletion.Hum Mutat. 2018 Apr;39(4):537-549. doi: 10.1002/humu.23396. Epub 2018 Jan 22. Hum Mutat. 2018. PMID: 29297947 Free PMC article.
-
Special delivery: distributing iron in the cytosol of mammalian cells.Front Pharmacol. 2014 Jul 22;5:173. doi: 10.3389/fphar.2014.00173. eCollection 2014. Front Pharmacol. 2014. PMID: 25101000 Free PMC article. Review.
References
-
- Al-shareef I, Arabi M, Dabbagh O. Cardiac involvement in nonketotic hyperglycinemia. J Child Neurol. 2011;26:970–3. - PubMed
-
- Bank WJ, Morrow G. A familial spinal cord disorder with hyperglycinemia. Arch Neurol. 1972;27:136–44. - PubMed
-
- Camaschella C, Campanella A, De Falco L, Boschetto L, Merlini R, Silvestri L, et al. The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload. Blood. 2007;110:1353–8. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
