

Novel dynein DYNC1H1 neck and motor domain mutations link distal spinal muscular atrophy and abnormal cortical development
- PMID: 24307404
- PMCID: PMC4109683
- DOI: 10.1002/humu.22491
Novel dynein DYNC1H1 neck and motor domain mutations link distal spinal muscular atrophy and abnormal cortical development
Abstract
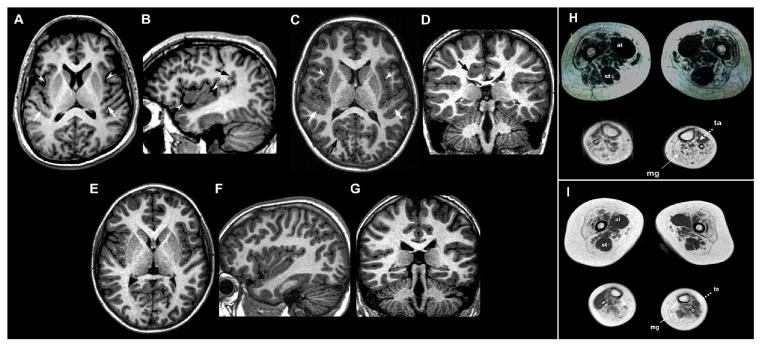
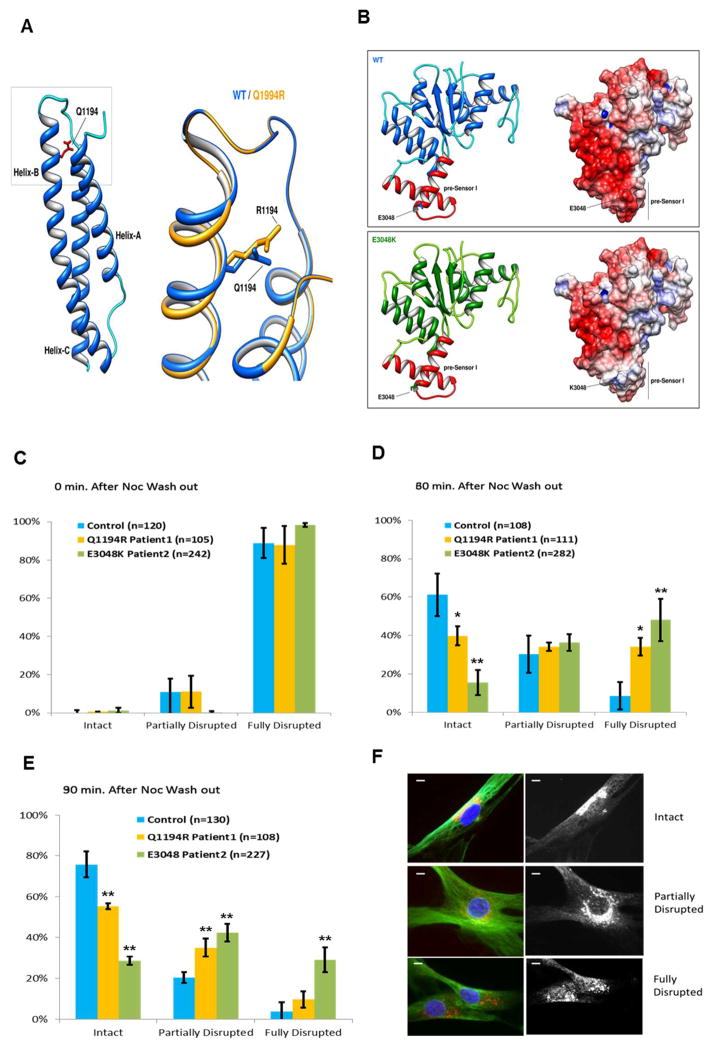
DYNC1H1 encodes the heavy chain of cytoplasmic dynein 1, a motor protein complex implicated in retrograde axonal transport, neuronal migration, and other intracellular motility functions. Mutations in DYNC1H1 have been described in autosomal-dominant Charcot-Marie-Tooth type 2 and in families with distal spinal muscular atrophy (SMA) predominantly affecting the legs (SMA-LED). Recently, defects of cytoplasmic dynein 1 were also associated with a form of mental retardation and neuronal migration disorders. Here, we describe two unrelated patients presenting a combined phenotype of congenital motor neuron disease associated with focal areas of cortical malformation. In each patient, we identified a novel de novo mutation in DYNC1H1: c.3581A>G (p.Gln1194Arg) in one case and c.9142G>A (p.Glu3048Lys) in the other. The mutations lie in different domains of the dynein heavy chain, and are deleterious to protein function as indicated by assays for Golgi recovery after nocodazole washout in patient fibroblasts. Our results expand the set of pathological mutations in DYNC1H1, reinforce the role of cytoplasmic dynein in disorders of neuronal migration, and provide evidence for a syndrome including spinal nerve degeneration and brain developmental problems.
Keywords: DYNC1H1; SMA-LED; abnormal cortical development; distal SMA.
© 2013 WILEY PERIODICALS, INC.
Conflict of interest statement
All authors declare no conflict of interest.
Figures
Similar articles
-
Novel mutations in the DYNC1H1 tail domain refine the genetic and clinical spectrum of dyneinopathies.Hum Mutat. 2015 Mar;36(3):287-91. doi: 10.1002/humu.22744. Hum Mutat. 2015. PMID: 25512093
-
Exome Sequencing Identifies DYNC1H1 Variant Associated With Vertebral Abnormality and Spinal Muscular Atrophy With Lower Extremity Predominance.Pediatr Neurol. 2015 Feb;52(2):239-44. doi: 10.1016/j.pediatrneurol.2014.09.003. Epub 2014 Oct 5. Pediatr Neurol. 2015. PMID: 25484024 Free PMC article.
-
A missense mutation in DYNC1H1 gene causing spinal muscular atrophy - Lower extremity, dominant.Neurol Neurochir Pol. 2018 Mar;52(2):293-297. doi: 10.1016/j.pjnns.2017.12.004. Epub 2017 Dec 14. Neurol Neurochir Pol. 2018. PMID: 29306600
-
Whole-exome sequencing identifies a novel de novo variant in DYNC1H in a patient with intractable epilepsy.Neurol Sci. 2022 Apr;43(4):2853-2858. doi: 10.1007/s10072-021-05824-9. Epub 2022 Jan 28. Neurol Sci. 2022. PMID: 35088241 Review.
-
Cytoplasmic dynein heavy chain: the servant of many masters.Trends Neurosci. 2013 Nov;36(11):641-51. doi: 10.1016/j.tins.2013.08.001. Epub 2013 Sep 10. Trends Neurosci. 2013. PMID: 24035135 Free PMC article. Review.
Cited by
-
Molecular basis for dyneinopathies reveals insight into dynein regulation and dysfunction.Elife. 2019 Jul 31;8:e47246. doi: 10.7554/eLife.47246. Elife. 2019. PMID: 31364990 Free PMC article.
-
Cytoplasmic dynein and its regulatory proteins in Golgi pathology in nervous system disorders.Front Neurosci. 2015 Oct 26;9:397. doi: 10.3389/fnins.2015.00397. eCollection 2015. Front Neurosci. 2015. PMID: 26578860 Free PMC article. Review.
-
Whole-exome sequencing identifies a novel de novo mutation in DYNC1H1 in epileptic encephalopathies.Sci Rep. 2017 Mar 21;7(1):258. doi: 10.1038/s41598-017-00208-6. Sci Rep. 2017. PMID: 28325891 Free PMC article.
-
Cargo specificity, regulation, and therapeutic potential of cytoplasmic dynein.Exp Mol Med. 2024 Apr;56(4):827-835. doi: 10.1038/s12276-024-01200-7. Epub 2024 Apr 1. Exp Mol Med. 2024. PMID: 38556551 Free PMC article. Review.
-
MRI patterns of muscle involvement in type 2 and 3 spinal muscular atrophy patients.J Neurol. 2020 Apr;267(4):898-912. doi: 10.1007/s00415-019-09646-w. Epub 2019 Nov 27. J Neurol. 2020. PMID: 31776722
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
