

De Novo mutations in GNAO1, encoding a Gαo subunit of heterotrimeric G proteins, cause epileptic encephalopathy
- PMID: 23993195
- PMCID: PMC3769919
- DOI: 10.1016/j.ajhg.2013.07.014
De Novo mutations in GNAO1, encoding a Gαo subunit of heterotrimeric G proteins, cause epileptic encephalopathy
Abstract
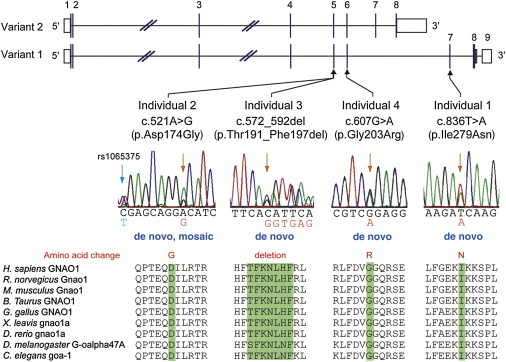
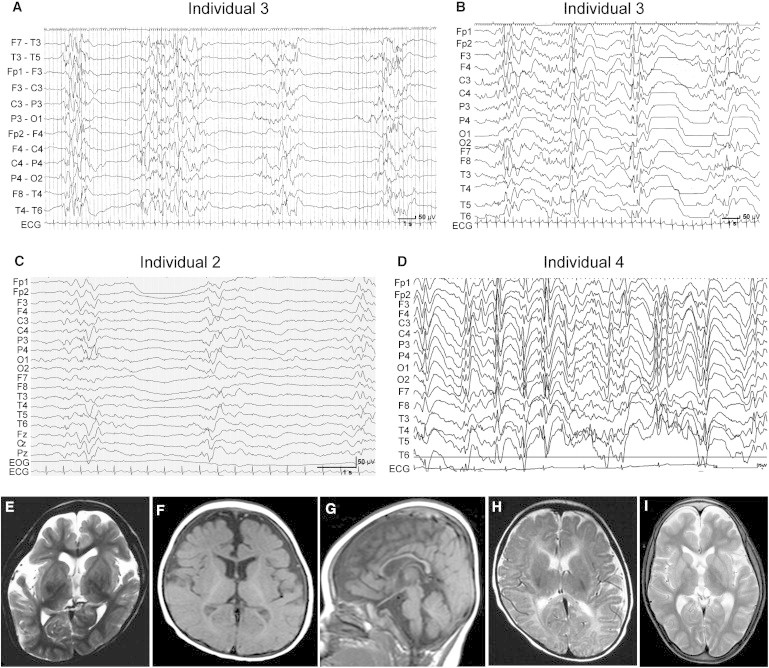
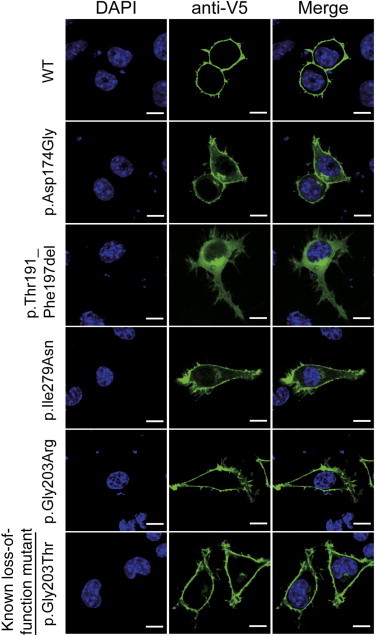
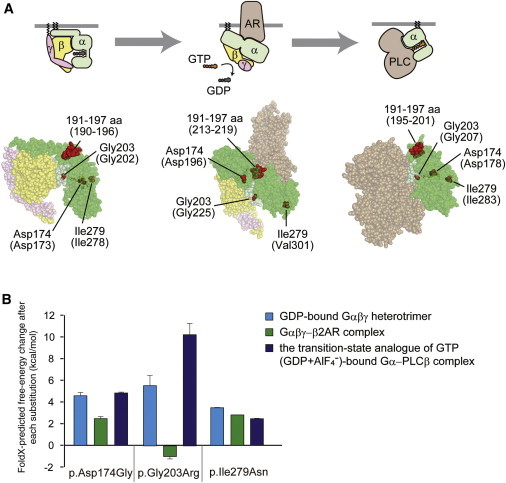
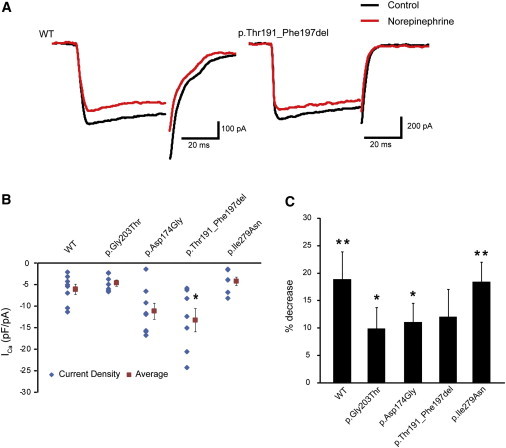
Heterotrimeric G proteins, composed of α, β, and γ subunits, can transduce a variety of signals from seven-transmembrane-type receptors to intracellular effectors. By whole-exome sequencing and subsequent mutation screening, we identified de novo heterozygous mutations in GNAO1, which encodes a Gαo subunit of heterotrimeric G proteins, in four individuals with epileptic encephalopathy. Two of the affected individuals also showed involuntary movements. Somatic mosaicism (approximately 35% to 50% of cells, distributed across multiple cell types, harbored the mutation) was shown in one individual. By mapping the mutation onto three-dimensional models of the Gα subunit in three different complexed states, we found that the three mutants (c.521A>G [p.Asp174Gly], c.836T>A [p.Ile279Asn], and c.572_592del [p.Thr191_Phe197del]) are predicted to destabilize the Gα subunit fold. A fourth mutant (c.607G>A), in which the Gly203 residue located within the highly conserved switch II region is substituted to Arg, is predicted to impair GTP binding and/or activation of downstream effectors, although the p.Gly203Arg substitution might not interfere with Gα binding to G-protein-coupled receptors. Transient-expression experiments suggested that localization to the plasma membrane was variably impaired in the three putatively destabilized mutants. Electrophysiological analysis showed that Gαo-mediated inhibition of calcium currents by norepinephrine tended to be lower in three of the four Gαo mutants. These data suggest that aberrant Gαo signaling can cause multiple neurodevelopmental phenotypes, including epileptic encephalopathy and involuntary movements.
Copyright © 2013 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Pediatric Encephalopathy: Clinical, Biochemical and Cellular Insights into the Role of Gln52 of GNAO1 and GNAI1 for the Dominant Disease.Cells. 2021 Oct 14;10(10):2749. doi: 10.3390/cells10102749. Cells. 2021. PMID: 34685729 Free PMC article.
-
Different biochemical properties explain why two equivalent Gα subunit mutants cause unrelated diseases.J Biol Chem. 2014 Aug 8;289(32):21818-27. doi: 10.1074/jbc.M114.549790. Epub 2014 Jun 30. J Biol Chem. 2014. PMID: 24982418 Free PMC article.
-
Molecular basis of a novel oncogenic mutation in GNAO1.Oncogene. 2011 Jun 9;30(23):2691-6. doi: 10.1038/onc.2010.645. Epub 2011 Feb 14. Oncogene. 2011. PMID: 21317923 Free PMC article.
-
A mechanistic review on GNAO1-associated movement disorder.Neurobiol Dis. 2018 Aug;116:131-141. doi: 10.1016/j.nbd.2018.05.005. Epub 2018 May 24. Neurobiol Dis. 2018. PMID: 29758257 Review.
-
Expanding Phenotype of De Novo Mutations in GNAO1: Four New Cases and Review of Literature.Neuropediatrics. 2017 Oct;48(5):371-377. doi: 10.1055/s-0037-1603977. Epub 2017 Jun 19. Neuropediatrics. 2017. PMID: 28628939 Review. No abstract available.
Cited by
-
Molecular annotation of G protein variants in a neurological disorder.Cell Rep. 2023 Dec 26;42(12):113462. doi: 10.1016/j.celrep.2023.113462. Epub 2023 Nov 18. Cell Rep. 2023. PMID: 37980565 Free PMC article.
-
GNAO1-related neurodevelopmental disorder: Literature review and caregiver survey.Epilepsy Behav Rep. 2022 Dec 31;21:100582. doi: 10.1016/j.ebr.2022.100582. eCollection 2023. Epilepsy Behav Rep. 2022. PMID: 36654732 Free PMC article.
-
Phenotypes in children with GNAO1 encephalopathy in China.Front Pediatr. 2023 Aug 29;11:1086970. doi: 10.3389/fped.2023.1086970. eCollection 2023. Front Pediatr. 2023. PMID: 37705601 Free PMC article.
-
Highlighting the Dystonic Phenotype Related to GNAO1.Mov Disord. 2022 Jul;37(7):1547-1554. doi: 10.1002/mds.29074. Epub 2022 Jun 20. Mov Disord. 2022. PMID: 35722775 Free PMC article.
-
Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions.Genet Med. 2015 Jul;17(7):578-86. doi: 10.1038/gim.2014.154. Epub 2014 Nov 13. Genet Med. 2015. PMID: 25356970
References
-
- Dulac O. Epileptic encephalopathy. Epilepsia. 2001;42(Suppl 3):23–26. - PubMed
-
- Ohtahara S., Yamatogi Y. Ohtahara syndrome: with special reference to its developmental aspects for differentiating from early myoclonic encephalopathy. Epilepsy Res. 2006;70(Suppl 1):S58–S67. - PubMed
-
- Saitsu H., Kato M., Koide A., Goto T., Fujita T., Nishiyama K., Tsurusaki Y., Doi H., Miyake N., Hayasaka K., Matsumoto N. Whole exome sequencing identifies KCNQ2 mutations in Ohtahara syndrome. Ann. Neurol. 2012;72:298–300. - PubMed
-
- Saitsu H., Kato M., Mizuguchi T., Hamada K., Osaka H., Tohyama J., Uruno K., Kumada S., Nishiyama K., Nishimura A. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat. Genet. 2008;40:782–788. - PubMed
-
- Weckhuysen S., Mandelstam S., Suls A., Audenaert D., Deconinck T., Claes L.R., Deprez L., Smets K., Hristova D., Yordanova I. KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann. Neurol. 2012;71:15–25. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
