

GRIN2A mutations cause epilepsy-aphasia spectrum disorders
- PMID: 23933818
- PMCID: PMC3868952
- DOI: 10.1038/ng.2727
GRIN2A mutations cause epilepsy-aphasia spectrum disorders
Abstract
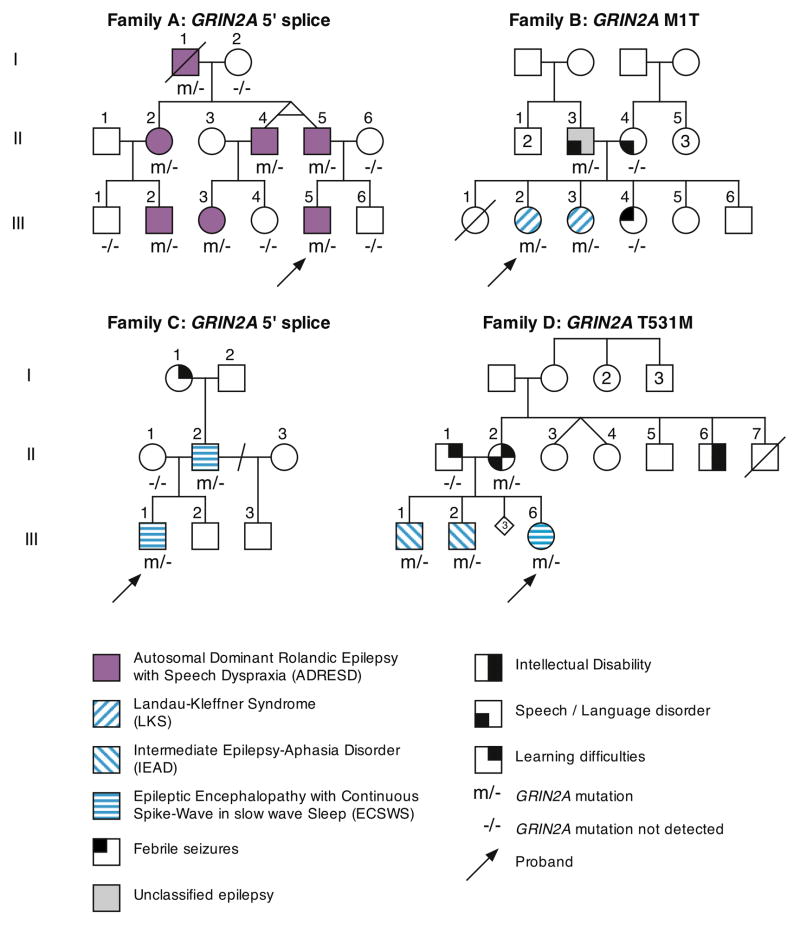
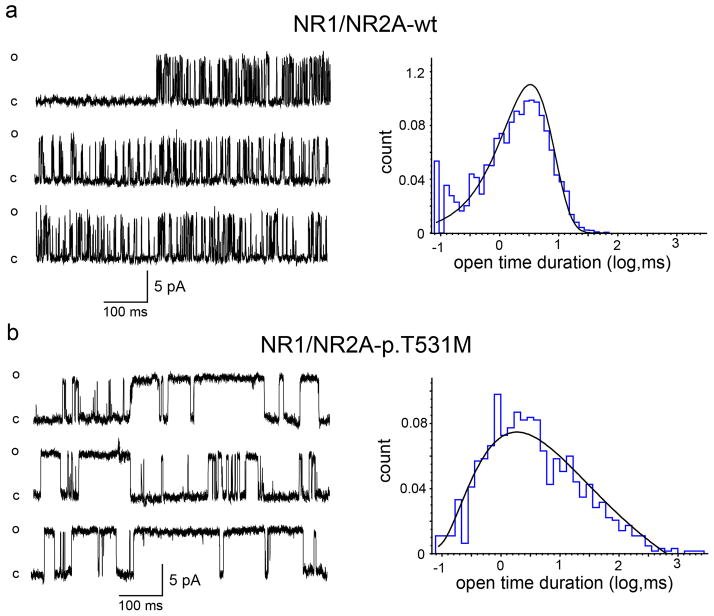
Epilepsy-aphasia syndromes (EAS) are a group of rare, severe epileptic encephalopathies of unknown etiology with a characteristic electroencephalogram (EEG) pattern and developmental regression particularly affecting language. Rare pathogenic deletions that include GRIN2A have been implicated in neurodevelopmental disorders. We sought to delineate the pathogenic role of GRIN2A in 519 probands with epileptic encephalopathies with diverse epilepsy syndromes. We identified four probands with GRIN2A variants that segregated with the disorder in their families. Notably, all four families presented with EAS, accounting for 9% of epilepsy-aphasia cases. We did not detect pathogenic variants in GRIN2A in other epileptic encephalopathies (n = 475) nor in probands with benign childhood epilepsy with centrotemporal spikes (n = 81). We report the first monogenic cause, to our knowledge, for EAS. GRIN2A mutations are restricted to this group of cases, which has important ramifications for diagnostic testing and treatment and provides new insights into the pathogenesis of this debilitating group of conditions.
Figures
Comment in
-
Epilepsy: GRIN2A mutations identified as key genetic drivers of epilepsy-aphasia spectrum disorders.Nat Rev Neurol. 2013 Oct;9(10):541. doi: 10.1038/nrneurol.2013.181. Epub 2013 Sep 3. Nat Rev Neurol. 2013. PMID: 23999465 No abstract available.
Similar articles
-
GRIN2A mutations in epilepsy-aphasia spectrum disorders.Brain Dev. 2018 Mar;40(3):205-210. doi: 10.1016/j.braindev.2017.09.007. Epub 2017 Oct 19. Brain Dev. 2018. PMID: 29056244
-
Update on the genetics of the epilepsy-aphasia spectrum and role of GRIN2A mutations.Epileptic Disord. 2019 Jun 1;21(S1):41-47. doi: 10.1684/epd.2019.1056. Epileptic Disord. 2019. PMID: 31149903 Review.
-
[Study of GRIN2A mutation in epilepsy-aphasia spectrum disorders].Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2018 Jun 10;35(3):314-318. doi: 10.3760/cma.j.issn.1003-9406.2018.03.002. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2018. PMID: 29896722 Chinese.
-
GRIN2A mutations in acquired epileptic aphasia and related childhood focal epilepsies and encephalopathies with speech and language dysfunction.Nat Genet. 2013 Sep;45(9):1061-6. doi: 10.1038/ng.2726. Epub 2013 Aug 11. Nat Genet. 2013. PMID: 23933820
-
GRIN2A-related epilepsy and speech disorders: A comprehensive overview with a focus on the role of precision therapeutics.Epilepsy Res. 2023 Jan;189:107065. doi: 10.1016/j.eplepsyres.2022.107065. Epub 2022 Dec 11. Epilepsy Res. 2023. PMID: 36516565 Review.
Cited by
-
Differential Functional Changes of Nav1.2 Channel Causing SCN2A-Related Epilepsy and Status Epilepticus During Slow Sleep.Front Neurol. 2021 May 19;12:653517. doi: 10.3389/fneur.2021.653517. eCollection 2021. Front Neurol. 2021. PMID: 34093402 Free PMC article.
-
GRIN2A mutation is a novel indicator of stratifying beneficiaries of immune checkpoint inhibitors in multiple cancers.Cancer Gene Ther. 2024 Apr;31(4):586-598. doi: 10.1038/s41417-024-00730-6. Epub 2024 Jan 24. Cancer Gene Ther. 2024. PMID: 38267623
-
Rare genetic brain disorders with overlapping neurological and psychiatric phenotypes.Nat Rev Neurol. 2024 Jan;20(1):7-21. doi: 10.1038/s41582-023-00896-x. Epub 2023 Nov 24. Nat Rev Neurol. 2024. PMID: 38001363 Review.
-
Common synaptic phenotypes arising from diverse mutations in the human NMDA receptor subunit GluN2A.Commun Biol. 2022 Feb 28;5(1):174. doi: 10.1038/s42003-022-03115-3. Commun Biol. 2022. PMID: 35228668 Free PMC article.
-
Imprecision in Precision Medicine: Differential Response of a Disease-Linked GluN2A Mutant to NMDA Channel Blockers.Front Pharmacol. 2021 Oct 28;12:773455. doi: 10.3389/fphar.2021.773455. eCollection 2021. Front Pharmacol. 2021. PMID: 34776984 Free PMC article.
References
-
- Berg AT, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia. 2010;51:676–85. - PubMed
-
- Tassinari CA, et al. Encephalopathy with electrical status epilepticus during slow sleep or ESES syndrome including the acquired aphasia. Clin Neurophysiol. 2000;111 (Suppl 2):S94–S102. - PubMed
-
- Tsai M, et al. Clinical genetic study of the epilepsy-aphasia spectrum. Epilepsia. 2012;54:280–287. - PubMed
-
- Deonna TW, Roulet E, Fontan D, Marcoz JP. Speech and oromotor deficits of epileptic origin in benign partial epilepsy of childhood with rolandic spikes (BPERS). Relationship to the acquired aphasia-epilepsy syndrome. Neuropediatrics. 1993;24:83–87. - PubMed
-
- Scheffer IE, et al. Autosomal dominant rolandic epilepsy and speech dyspraxia: a new syndrome with anticipation. Ann Neurol. 1995;38:633–642. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
