

Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1
- PMID: 23708187
- PMCID: PMC3704157
- DOI: 10.1038/ng.2646
Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1
Abstract
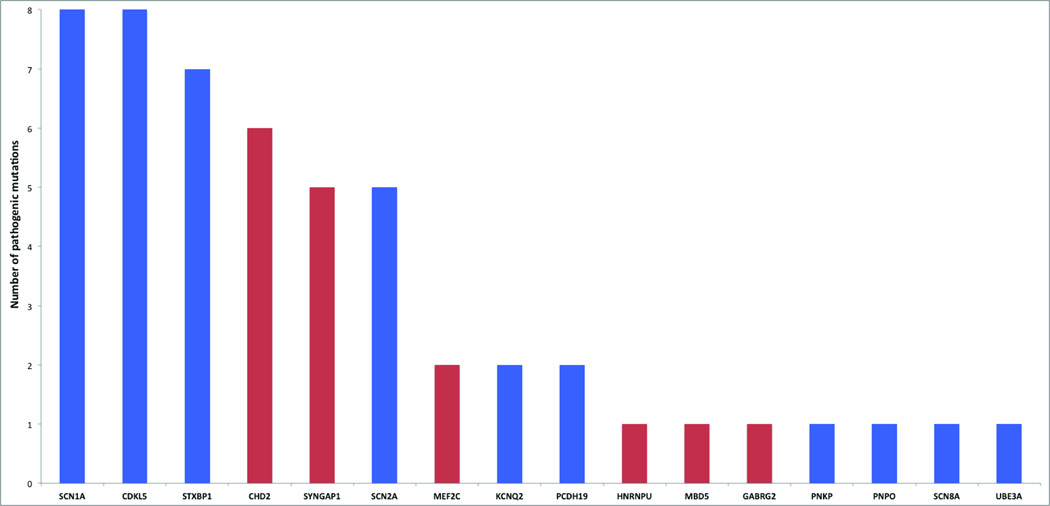
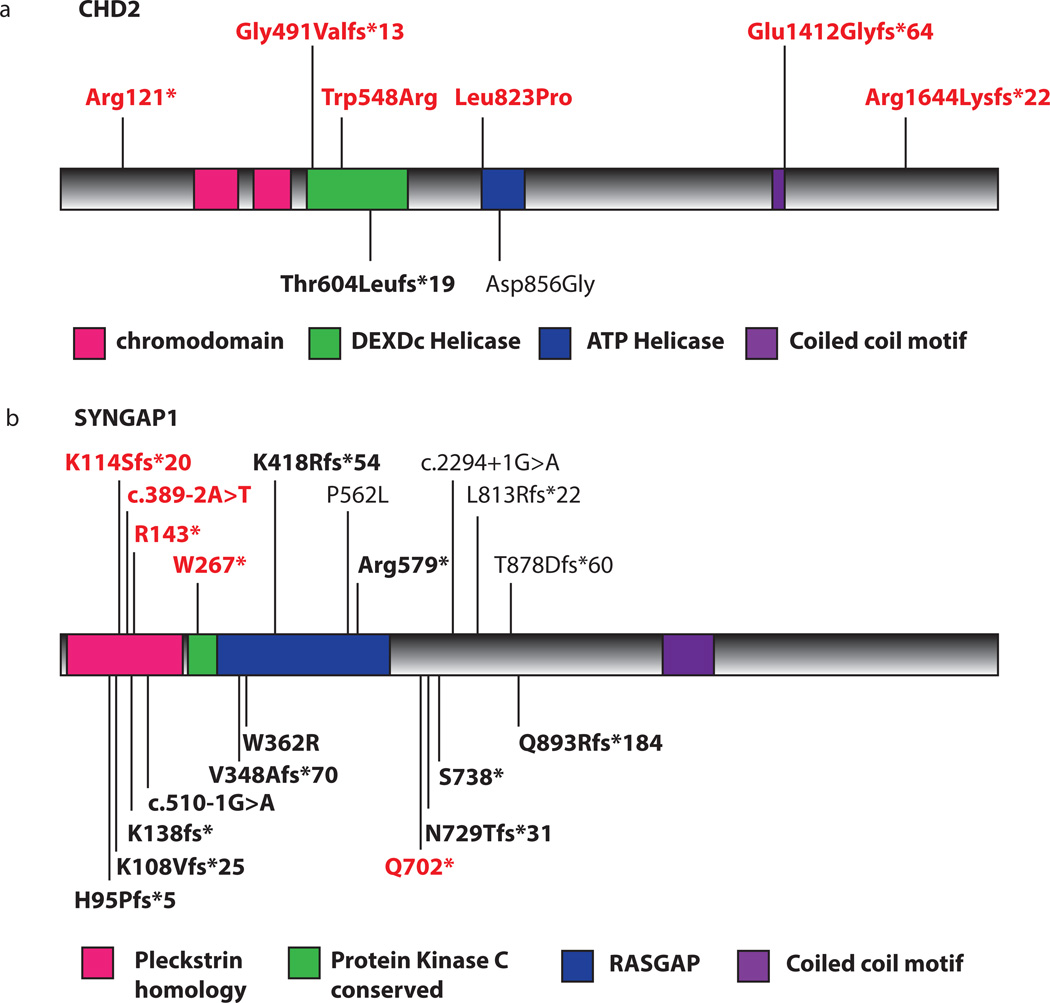
Epileptic encephalopathies are a devastating group of epilepsies with poor prognosis, of which the majority are of unknown etiology. We perform targeted massively parallel resequencing of 19 known and 46 candidate genes for epileptic encephalopathy in 500 affected individuals (cases) to identify new genes involved and to investigate the phenotypic spectrum associated with mutations in known genes. Overall, we identified pathogenic mutations in 10% of our cohort. Six of the 46 candidate genes had 1 or more pathogenic variants, collectively accounting for 3% of our cohort. We show that de novo CHD2 and SYNGAP1 mutations are new causes of epileptic encephalopathies, accounting for 1.2% and 1% of cases, respectively. We also expand the phenotypic spectra explained by SCN1A, SCN2A and SCN8A mutations. To our knowledge, this is the largest cohort of cases with epileptic encephalopathies to undergo targeted resequencing. Implementation of this rapid and efficient method will change diagnosis and understanding of the molecular etiologies of these disorders.
Figures
Similar articles
-
Novel mutations and phenotypes of epilepsy-associated genes in epileptic encephalopathies.Genes Brain Behav. 2018 Nov;17(8):e12456. doi: 10.1111/gbb.12456. Epub 2018 Jan 26. Genes Brain Behav. 2018. PMID: 29314583
-
KCNT1-related epilepsies and epileptic encephalopathies: phenotypic and mutational spectrum.Brain. 2021 Dec 31;144(12):3635-3650. doi: 10.1093/brain/awab219. Brain. 2021. PMID: 34114611
-
Exome sequencing reveals new causal mutations in children with epileptic encephalopathies.Epilepsia. 2013 Jul;54(7):1270-81. doi: 10.1111/epi.12201. Epub 2013 May 3. Epilepsia. 2013. PMID: 23647072 Free PMC article.
-
CHD2-Related CNS Pathologies.Int J Mol Sci. 2021 Jan 8;22(2):588. doi: 10.3390/ijms22020588. Int J Mol Sci. 2021. PMID: 33435571 Free PMC article. Review.
-
Chewing induced reflex seizures ("eating epilepsy") and eye closure sensitivity as a common feature in pediatric patients with SYNGAP1 mutations: Review of literature and report of 8 cases.Seizure. 2019 Feb;65:131-137. doi: 10.1016/j.seizure.2018.12.020. Epub 2018 Dec 22. Seizure. 2019. PMID: 30685520 Review.
Cited by
-
Mammalian Resilience Revealed by a Comparison of Human Diseases and Mouse Models Associated With DNA Helicase Deficiencies.Front Mol Biosci. 2022 Aug 11;9:934042. doi: 10.3389/fmolb.2022.934042. eCollection 2022. Front Mol Biosci. 2022. PMID: 36032672 Free PMC article. Review.
-
Voltage Gated Sodium Channel Genes in Epilepsy: Mutations, Functional Studies, and Treatment Dimensions.Front Neurol. 2021 Mar 24;12:600050. doi: 10.3389/fneur.2021.600050. eCollection 2021. Front Neurol. 2021. PMID: 33841294 Free PMC article. Review.
-
Targeted sequencing of 351 candidate genes for epileptic encephalopathy in a large cohort of patients.Mol Genet Genomic Med. 2016 Jul 30;4(5):568-80. doi: 10.1002/mgg3.235. eCollection 2016 Sep. Mol Genet Genomic Med. 2016. PMID: 27652284 Free PMC article.
-
Epileptic spasms are a feature of DEPDC5 mTORopathy.Neurol Genet. 2015 Jul 23;1(2):e17. doi: 10.1212/NXG.0000000000000016. eCollection 2015 Aug. Neurol Genet. 2015. PMID: 27066554 Free PMC article.
-
Molecular pathophysiology and pharmacology of the voltage-sensing module of neuronal ion channels.Front Cell Neurosci. 2015 Jul 15;9:259. doi: 10.3389/fncel.2015.00259. eCollection 2015. Front Cell Neurosci. 2015. PMID: 26236192 Free PMC article. Review.
References
-
- Berg AT, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology 2005–2009. Epilepsia. 2010;51:676–685. - PubMed
-
- Kamien BA, Cardamone M, Lawson JA, Sachdev R. A genetic diagnostic approach to infantile epileptic encephalopathies. J. Clin. Neurosci. 2012;19:934–941. - PubMed
-
- Capelli LP, et al. Deletion of the RMGA and CHD2 genes in a child with epilepsy and mental deficiency. Eur. J. Med. Genet. 2012;55:132–134. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
