

Hereditary causes of kidney stones and chronic kidney disease
- PMID: 23334384
- PMCID: PMC4138059
- DOI: 10.1007/s00467-012-2329-z
Hereditary causes of kidney stones and chronic kidney disease
Abstract
Adenine phosphoribosyltransferase (APRT) deficiency, cystinuria, Dent disease, familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC), and primary hyperoxaluria (PH) are rare but important causes of severe kidney stone disease and/or chronic kidney disease in children. Recurrent kidney stone disease and nephrocalcinosis, particularly in pre-pubertal children, should alert the physician to the possibility of an inborn error of metabolism as the underlying cause. Unfortunately, the lack of recognition and knowledge of the five disorders has frequently resulted in an unacceptable delay in diagnosis and treatment, sometimes with grave consequences. A high index of suspicion coupled with early diagnosis may reduce or even prevent the serious long-term complications of these diseases. In this paper, we review the epidemiology, clinical features, diagnosis, treatment, and outcome of patients with APRT deficiency, cystinuria, Dent disease, FHHNC, and PH, with an emphasis on childhood manifestations.
Conflict of interest statement
Figures
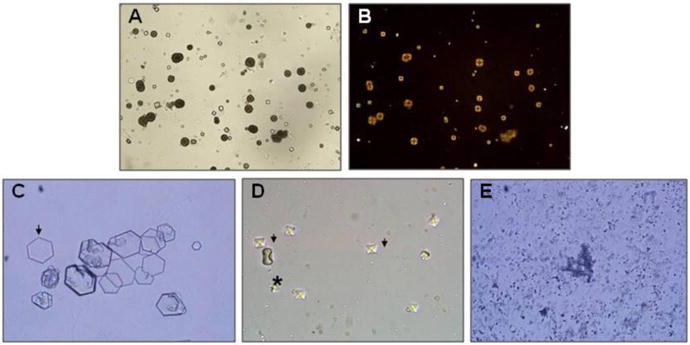
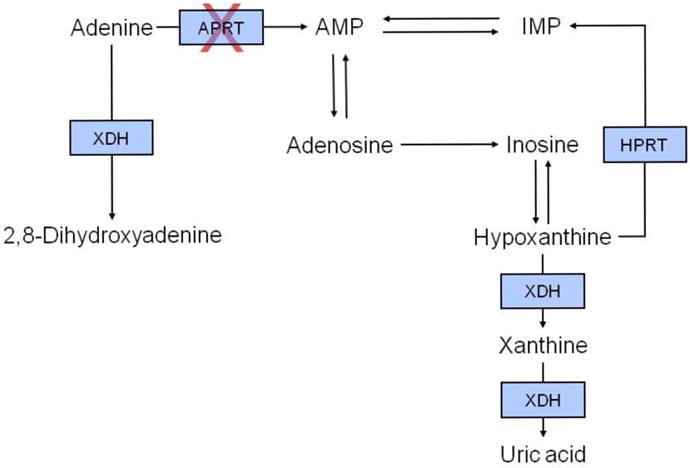
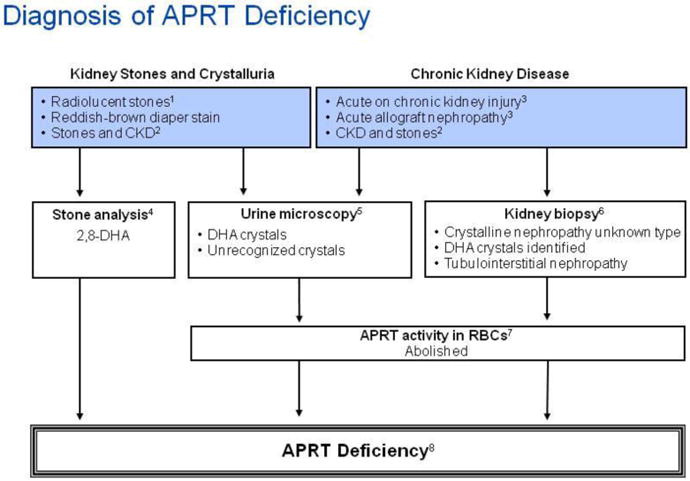
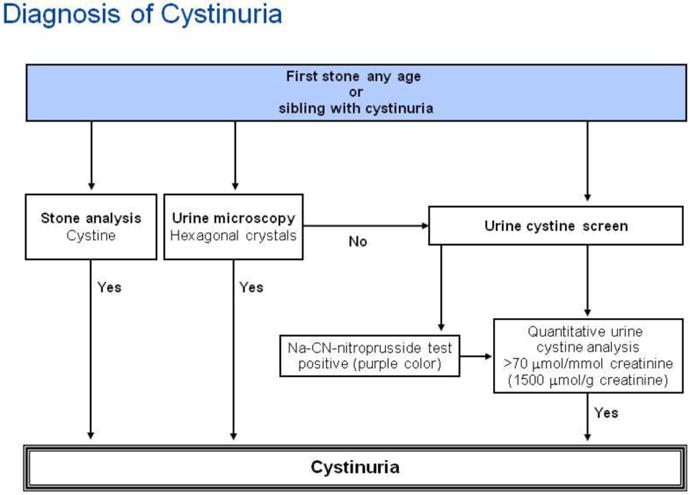
| Random urine calcium-to-creatinine ratio (Ca/Cr) by age [119] | ||
| Age (yr) | Ca/Cr ratio Upper limit of normal | |
| (mmol/mmol) | (mg/mg) | |
| 0-1 | 2.29 | 0.81 |
| 1-2 | 1.58 | 0.56 |
| 2-3 | 1.41 | 0.50 |
| 3-5 | 1.16 | 0.41 |
| 5-7 | 0.85 | 0.30 |
| 7-10 | 0.71 | 0.25 |
| 10-14 | 0.68 | 0.24 |
| 14-17 | 0.68 | 0.24 |
| Random urine magnesium-to-creatinine (Mg/Cr) ratio by age [119] | ||
| Age (yr) | Mg/Cr ratioUpper limit of normal | |
| mmo/mmol | mg/mg | |
| 0-1 | 2.2 | 0.48 |
| 1-2 | 1.7 | 0.37 |
| 2-3 | 1.6 | 0.34 |
| 3-5 | 1.3 | 0.29 |
| 5-7 | 1.0 | 0.21 |
| 7-10 | 0.9 | 0.18 |
| 10-14 | 0.7 | 0.15 |
| 14-17 | 0.6 | 0.13 |
| Random urine citrate-to-creatinine (Cit/Cr) ratio by age [120] | ||
| Age (yr) | Cit/Cr ratio Lower limit of normal | |
| mmol/mmol | mg/mg | |
| 0-5 | 0.25 | 0.42 |
| >5 | 0.15 | 0.25 |
| Random urine oxalate-to-creatinine (Ox/Cr) ratio by age [123-126] | ||
| Age | Ox/Cr ratio Upper limit of normal | |
| (mmol/mmol) | (mg/mg) | |
| <6 months | 0.37 | 0.29 |
| 6 months to 2 years | 0.26 | 0.20 |
| >2 years to 5 years | 0.14 | 0.11 |
| 6 to 12 years | 0.08 | 0.063 |
Similar articles
-
Early Recognition and Management of Rare Kidney Stone Disorders.Urol Nurs. 2017 Mar-Apr;37(2):81-9, 102. Urol Nurs. 2017. PMID: 29240373 Free PMC article.
-
Renal stones in two children with two rare etiologies.Saudi J Kidney Dis Transpl. 2018 Sep-Oct;29(5):1203-1206. doi: 10.4103/1319-2442.243955. Saudi J Kidney Dis Transpl. 2018. PMID: 30381520
-
Genetic defects underlying renal stone disease.Int J Surg. 2016 Dec;36(Pt D):590-595. doi: 10.1016/j.ijsu.2016.11.015. Epub 2016 Nov 10. Int J Surg. 2016. PMID: 27838384 Review.
-
Research progress on renal calculus associate with inborn error of metabolism.Zhejiang Da Xue Xue Bao Yi Xue Ban. 2023 Apr 25;52(2):169-177. doi: 10.3724/zdxbyxb-2022-0698. Zhejiang Da Xue Xue Bao Yi Xue Ban. 2023. PMID: 37283101 Free PMC article. Review. Chinese, English.
-
Familial hypomagnesemia with hypercalciuria and nephrocalcinosis: unusual clinical associations and novel claudin16 mutation in an Egyptian family.Clin Exp Nephrol. 2009 Aug;13(4):288-294. doi: 10.1007/s10157-008-0126-6. Epub 2009 Jan 24. Clin Exp Nephrol. 2009. PMID: 19165416
Cited by
-
A Rare Sparkle: A Case of Calcified Kidneys in a Young Infant With Renal Failure.Cureus. 2023 Oct 11;15(10):e46827. doi: 10.7759/cureus.46827. eCollection 2023 Oct. Cureus. 2023. PMID: 37954792 Free PMC article.
-
Metabolic diagnosis and medical prevention of calcium nephrolithiasis and its systemic manifestations: a consensus statement.J Nephrol. 2016 Dec;29(6):715-734. doi: 10.1007/s40620-016-0329-y. Epub 2016 Jul 25. J Nephrol. 2016. PMID: 27456839 Free PMC article. Review.
-
Dent Disease in Chinese Children and Findings from Heterozygous Mothers: Phenotypic Heterogeneity, Fetal Growth, and 10 Novel Mutations.J Pediatr. 2016 Jul;174:204-210.e1. doi: 10.1016/j.jpeds.2016.04.007. Epub 2016 May 9. J Pediatr. 2016. PMID: 27174143 Free PMC article.
-
HPO2Vec+: Leveraging heterogeneous knowledge resources to enrich node embeddings for the Human Phenotype Ontology.J Biomed Inform. 2019 Aug;96:103246. doi: 10.1016/j.jbi.2019.103246. Epub 2019 Jun 27. J Biomed Inform. 2019. PMID: 31255713 Free PMC article.
-
The Clinical Impact of Accurate Cystine Calculi Characterization Using Dual-Energy Computed Tomography.Case Rep Radiol. 2015;2015:801021. doi: 10.1155/2015/801021. Epub 2015 Nov 25. Case Rep Radiol. 2015. PMID: 26688770 Free PMC article.
References
-
- Sas DJ. An update on the changing epidemiology and metabolic risk factors in pediatric kidney stone disease. Clin J Am Soc Nephrol. 2011;6:2062–2068. - PubMed
-
- Stamatelou KK, Francis ME, Jones CA, Nyberg LM, Curhan GC. Time trends in reported prevalence of kidney stones in the United States: 1976-1994. Kidney Int. 2003;63:1817–1823. - PubMed
-
- Beara-Lasic L, Edvardsson V, Palsson R, Lieske J, Goldfarb D, Milliner D. Genetic causes of kidney stones and kidney failure. Clin Rev Bone Miner Metab. 2011;10(6):1–18.
-
- Greenwood MC, Dillon MJ, Simmonds HA, Barratt TM, Pincott JR, Metreweli C. Renal failure due to 2,8-dihydroxyadenine urolithiasis. Eur J Pediatr. 1982;138:346–349. - PubMed
-
- Konrad M, Hou J, Weber S, Dotsch J, Kari JA, Seeman T, Kuwertz-Broking E, Peco-Antic A, Tasic V, Dittrich K, Alshaya HO, von Vigier RO, Gallati S, Goodenough DA, Schaller A. CLDN16 genotype predicts renal decline in familial hypomagnesemia with hypercalciuria and nephrocalcinosis. J Am Soc Nephrol. 2008;19:171–181. - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
