

Genetic studies provide clues on the pathogenesis of idiopathic pulmonary fibrosis
- PMID: 23268535
- PMCID: PMC3529334
- DOI: 10.1242/dmm.010736
Genetic studies provide clues on the pathogenesis of idiopathic pulmonary fibrosis
Abstract
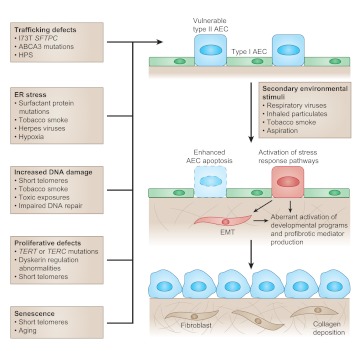
Idiopathic pulmonary fibrosis (IPF) is a progressive and often fatal lung disease for which there is no known treatment. Although the traditional paradigm of IPF pathogenesis emphasized chronic inflammation as the primary driver of fibrotic remodeling, more recent insights have challenged this view. Linkage analysis and candidate gene approaches have identified four genes that cause the inherited form of IPF, familial interstitial pneumonia (FIP). These four genes encode two surfactant proteins, surfactant protein C (encoded by SFTPC) and surfactant protein A2 (SFTPA2), and two components of the telomerase complex, telomerase reverse transcriptase (TERT) and the RNA component of telomerase (TERC). In this review, we discuss how investigating these mutations, as well as genetic variants identified in other inherited disorders associated with pulmonary fibrosis, are providing new insights into the pathogenesis of common idiopathic interstitial lung diseases, particularly IPF. Studies in this area have highlighted key roles for epithelial cell injury and dysfunction in the development of lung fibrosis. In addition, genetic approaches have uncovered the importance of several processes - including endoplasmic reticulum stress and the unfolded protein response, DNA-damage and -repair pathways, and cellular senescence - that might provide new therapeutic targets in fibrotic lung diseases.
Figures
Similar articles
-
Idiopathic pulmonary fibrosis: update on genetic discoveries.Proc Am Thorac Soc. 2011 May;8(2):158-62. doi: 10.1513/pats.201008-056MS. Proc Am Thorac Soc. 2011. PMID: 21543794 Free PMC article.
-
Genetics in pulmonary fibrosis--familial cases provide clues to the pathogenesis of idiopathic pulmonary fibrosis.Am J Med Sci. 2011 Jun;341(6):439-43. doi: 10.1097/MAJ.0b013e31821a9d7a. Am J Med Sci. 2011. PMID: 21613931 Free PMC article.
-
Molecular mechanisms in progressive idiopathic pulmonary fibrosis.Annu Rev Med. 2013;64:265-76. doi: 10.1146/annurev-med-042711-142004. Epub 2012 Sep 27. Annu Rev Med. 2013. PMID: 23020878 Review.
-
Resequencing Study Confirms That Host Defense and Cell Senescence Gene Variants Contribute to the Risk of Idiopathic Pulmonary Fibrosis.Am J Respir Crit Care Med. 2019 Jul 15;200(2):199-208. doi: 10.1164/rccm.201810-1891OC. Am J Respir Crit Care Med. 2019. PMID: 31034279 Free PMC article.
-
Familial forms of nonspecific interstitial pneumonia/idiopathic pulmonary fibrosis: clinical course and genetic background.Curr Opin Pulm Med. 2012 Sep;18(5):455-61. doi: 10.1097/MCP.0b013e328356b15c. Curr Opin Pulm Med. 2012. PMID: 22781209 Review.
Cited by
-
Interpretation of HRCT Scans in the Diagnosis of IPF: Improving Communication Between Pulmonologists and Radiologists.Lung. 2018 Oct;196(5):561-567. doi: 10.1007/s00408-018-0143-5. Epub 2018 Aug 10. Lung. 2018. PMID: 30097721 Free PMC article. Review.
-
Mouse Lung Fibroblast Resistance to Fas-Mediated Apoptosis Is Dependent on the Baculoviral Inhibitor of Apoptosis Protein 4 and the Cellular FLICE-Inhibitory Protein.Front Physiol. 2017 Mar 14;8:128. doi: 10.3389/fphys.2017.00128. eCollection 2017. Front Physiol. 2017. PMID: 28352235 Free PMC article.
-
Idiopathic Pulmonary Fibrosis: A Genetic Disease That Involves Mucociliary Dysfunction of the Peripheral Airways.Physiol Rev. 2016 Oct;96(4):1567-91. doi: 10.1152/physrev.00004.2016. Physiol Rev. 2016. PMID: 27630174 Free PMC article. Review.
-
Bone marrow mesenchymal stromal cells attenuate silica-induced pulmonary fibrosis potentially by attenuating Wnt/β-catenin signaling in rats.Stem Cell Res Ther. 2018 Nov 14;9(1):311. doi: 10.1186/s13287-018-1045-4. Stem Cell Res Ther. 2018. PMID: 30428918 Free PMC article.
-
A novel dyskerin (DKC1) mutation is associated with familial interstitial pneumonia.Chest. 2014 Jul;146(1):e1-e7. doi: 10.1378/chest.13-2224. Chest. 2014. PMID: 24504062 Free PMC article.
References
-
- Amin R. S., Wert S. E., Baughman R. P., Tomashefski J. F., Jr, Nogee L. M., Brody A. S., Hull W. M., Whitsett J. A. (2001). Surfactant protein deficiency in familial interstitial lung disease. J. Pediatr. 139, 85–92 - PubMed
-
- Aoshiba K., Tsuji T., Nagai A. (2003). Bleomycin induces cellular senescence in alveolar epithelial cells. Eur. Respir. J. 22, 436–443 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
