

Refractory epilepsy and mitochondrial dysfunction due to GM3 synthase deficiency
- PMID: 22990144
- PMCID: PMC3641379
- DOI: 10.1038/ejhg.2012.202
Refractory epilepsy and mitochondrial dysfunction due to GM3 synthase deficiency
Abstract
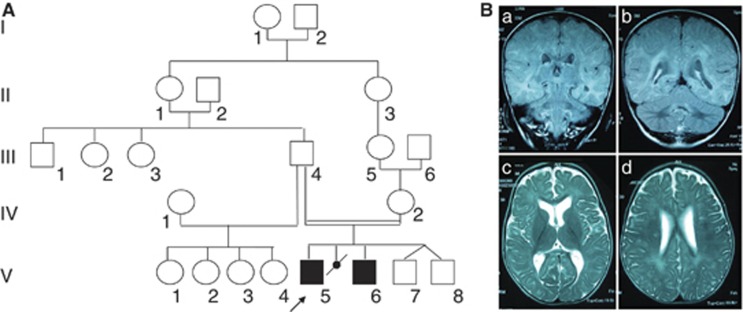
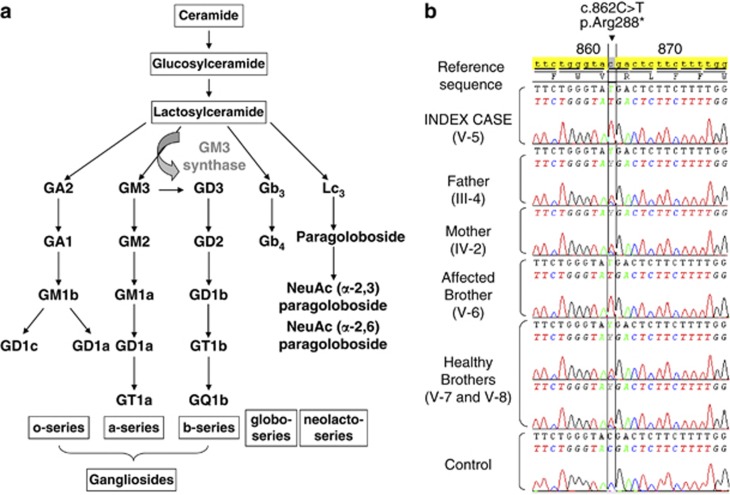
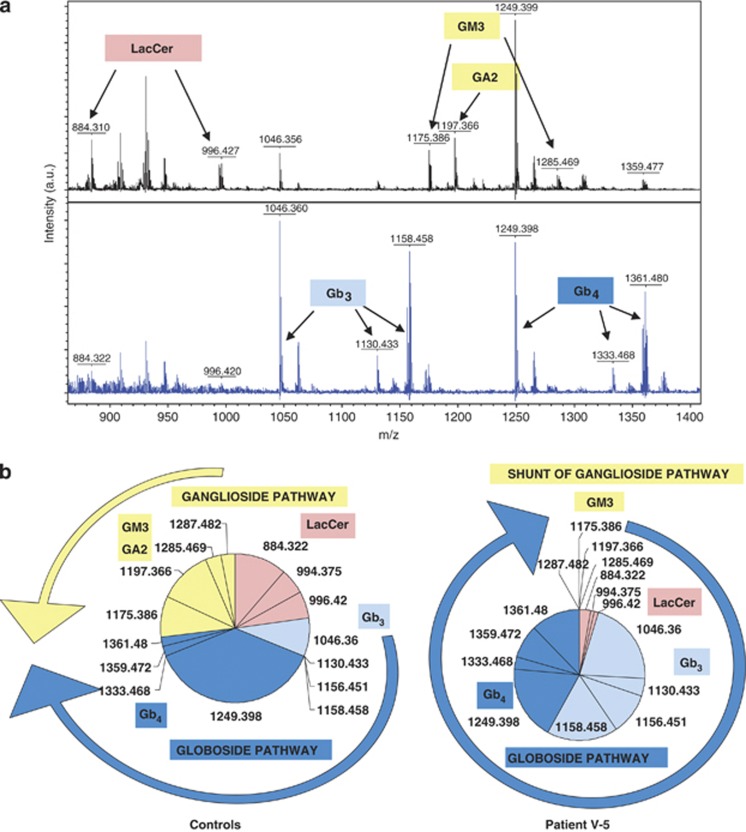
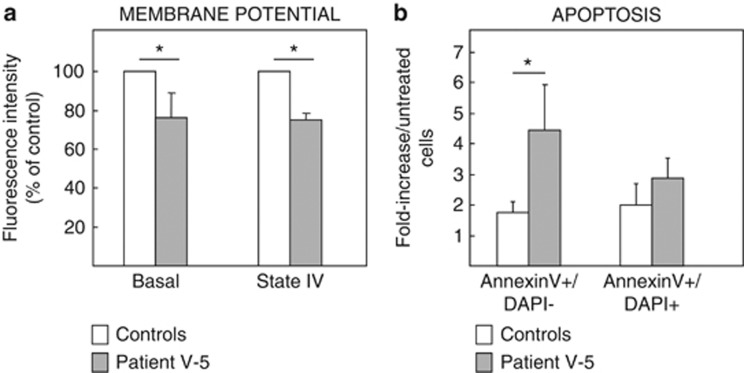
We report two children, born from consanguineous parents, who presented with early-onset refractory epilepsy associated with psychomotor delay, failure to thrive, blindness and deafness. Polarographic and spectrophotometric analyses in fibroblasts and liver revealed a respiratory chain (RC) dysfunction. Surprisingly, we identified a homozygous nonsense mutation in the GM3 synthase gene by using exome sequencing. GM3 synthase catalyzes the formation of GM3 ganglioside from lactosylceramide, which is the first step in the synthesis of complex ganglioside species. Mass spectrometry analysis revealed that the complete absence of GM3 ganglioside and its biosynthetic derivatives was associated with an upregulation of the alternative globoside pathway in fibroblasts. The accumulation of Gb3 and Gb4 globosides likely has a role in RC dysfunction and in the decrease of mitochondrial membrane potential leading to apoptosis, which we observed in fibroblasts. We show for the first time that GM3 synthase deficiency, responsible for early-onset epilepsy syndrome, leads to a secondary RC dysfunction. Our study highlights the role of secondary mitochondrial disorders that can interfere with the diagnosis and the evolution of other metabolic diseases.
Figures
Similar articles
-
Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-of-function mutation of GM3 synthase.Nat Genet. 2004 Nov;36(11):1225-9. doi: 10.1038/ng1460. Epub 2004 Oct 24. Nat Genet. 2004. PMID: 15502825
-
GM3 synthase deficiency due to ST3GAL5 variants in two Korean female siblings: Masquerading as Rett syndrome-like phenotype.Am J Med Genet A. 2016 Aug;170(8):2200-5. doi: 10.1002/ajmg.a.37773. Epub 2016 May 27. Am J Med Genet A. 2016. PMID: 27232954
-
HIBCH deficiency in a patient with phenotypic characteristics of mitochondrial disorders.Am J Med Genet A. 2014 Dec;164A(12):3162-9. doi: 10.1002/ajmg.a.36766. Epub 2014 Sep 23. Am J Med Genet A. 2014. PMID: 25251209
-
Ganglioside GM3 Synthase Deficiency in Mouse Models and Human Patients.Int J Mol Sci. 2022 May 11;23(10):5368. doi: 10.3390/ijms23105368. Int J Mol Sci. 2022. PMID: 35628171 Free PMC article. Review.
-
Phenotypic variation of TTC19-deficient mitochondrial complex III deficiency: a case report and literature review.Am J Med Genet A. 2015 Jun;167(6):1330-6. doi: 10.1002/ajmg.a.36968. Epub 2015 Apr 21. Am J Med Genet A. 2015. PMID: 25899669 Review.
Cited by
-
Enhanced Susceptibility to Chemoconvulsant-Induced Seizures in Ganglioside GM3 Synthase Knockout Mice.ASN Neuro. 2020 Jan-Dec;12:1759091420938175. doi: 10.1177/1759091420938175. ASN Neuro. 2020. PMID: 32664815 Free PMC article.
-
DEGS1-associated aberrant sphingolipid metabolism impairs nervous system function in humans.J Clin Invest. 2019 Mar 1;129(3):1229-1239. doi: 10.1172/JCI124159. Epub 2019 Feb 11. J Clin Invest. 2019. PMID: 30620338 Free PMC article.
-
Dystonia Due to GM3 Synthase Deficiency.Mov Disord Clin Pract. 2022 Jan 5;9(2):236-239. doi: 10.1002/mdc3.13399. eCollection 2022 Feb. Mov Disord Clin Pract. 2022. PMID: 35146061 Free PMC article.
-
Gangliosidome of a Human Hippocampus in Temporal Lobe Epilepsy Resolved by High-Resolution Tandem Mass Spectrometry.Molecules. 2022 Jun 23;27(13):4056. doi: 10.3390/molecules27134056. Molecules. 2022. PMID: 35807302 Free PMC article.
-
Start Me Up: How Can Surrounding Gangliosides Affect Sodium-Potassium ATPase Activity and Steer towards Pathological Ion Imbalance in Neurons?Biomedicines. 2022 Jun 27;10(7):1518. doi: 10.3390/biomedicines10071518. Biomedicines. 2022. PMID: 35884824 Free PMC article. Review.
References
-
- Debray F, Lambert M, Chevalier I, et al. Long-term outcome and clinical spectrum of 73 pediatric patients with mitochondrial diseases. Pediatrics. 2007;119:722–733. - PubMed
-
- Khurana D, Salganicoff L, Melvin J, et al. Epilepsy and respiratory chain defects in children with mitochondrial encephalopathy. Neuropediatrics. 2008;39:8–13. - PubMed
-
- Canafoglia L, Francheschetti S, Antozzi C, et al. Epileptic phenotypes associated with mitochondrial disorders. Neurology. 2001;56:1340–1346. - PubMed
-
- Lee Y, Kang H, Lee J, et al. Mitochondrial respiratory chain defects: underlying etiology in various epileptic conditions. Epilepsia. 2008;44:701–707. - PubMed
-
- El Sabbagh S, Lebre A, Bahi-Buisson N, et al. Epileptic phenotypes in children with respiratory chain disorders. Epilepsia. 2010;51:1225–1235. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
