

Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing
- PMID: 22277967
- PMCID: PMC3523805
- DOI: 10.1126/scitranslmed.3003310
Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing
Abstract
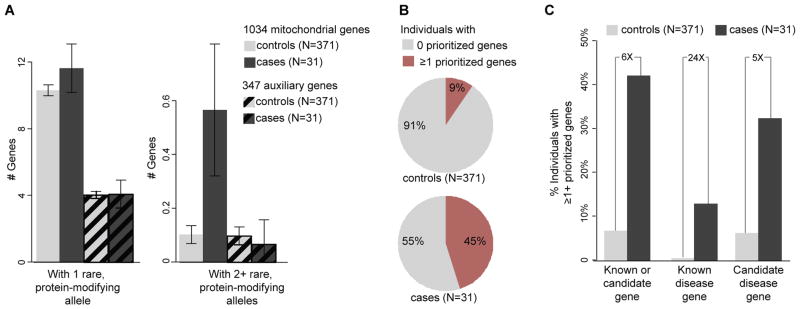
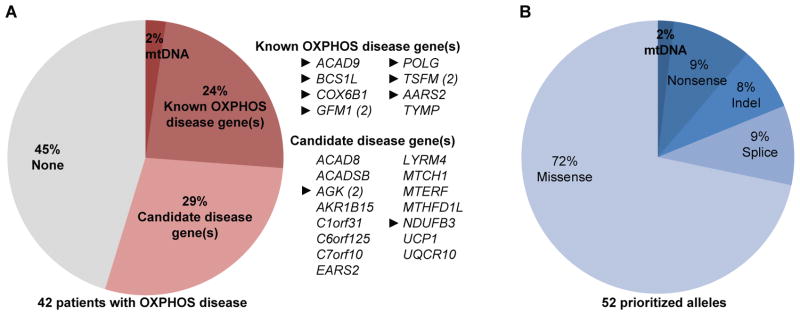
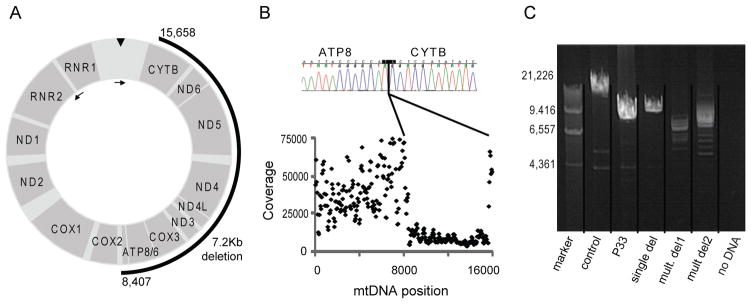
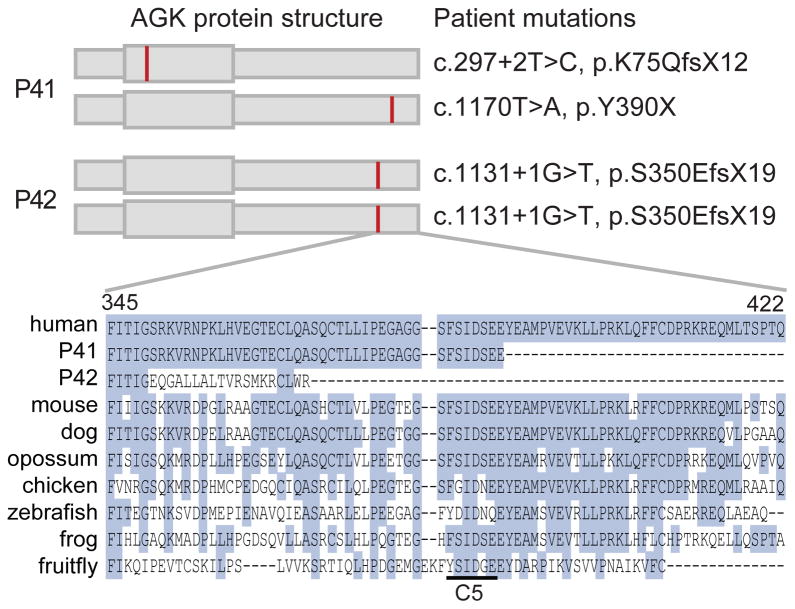
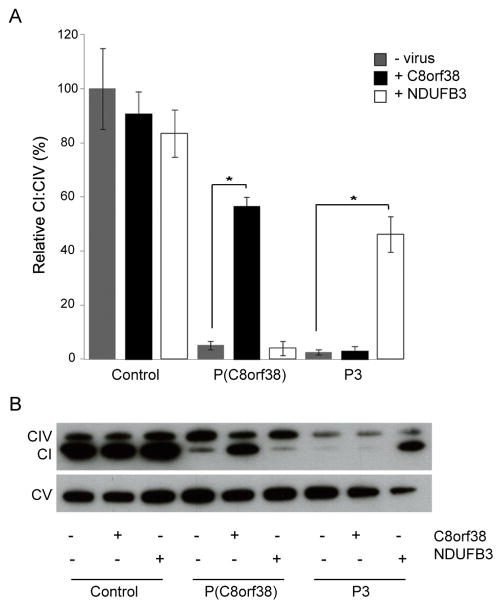
Advances in next-generation sequencing (NGS) promise to facilitate diagnosis of inherited disorders. Although in research settings NGS has pinpointed causal alleles using segregation in large families, the key challenge for clinical diagnosis is application to single individuals. To explore its diagnostic use, we performed targeted NGS in 42 unrelated infants with clinical and biochemical evidence of mitochondrial oxidative phosphorylation disease. These devastating mitochondrial disorders are characterized by phenotypic and genetic heterogeneity, with more than 100 causal genes identified to date. We performed "MitoExome" sequencing of the mitochondrial DNA (mtDNA) and exons of ~1000 nuclear genes encoding mitochondrial proteins and prioritized rare mutations predicted to disrupt function. Because patients and healthy control individuals harbored a comparable number of such heterozygous alleles, we could not prioritize dominant-acting genes. However, patients showed a fivefold enrichment of genes with two such mutations that could underlie recessive disease. In total, 23 of 42 (55%) patients harbored such recessive genes or pathogenic mtDNA variants. Firm diagnoses were enabled in 10 patients (24%) who had mutations in genes previously linked to disease. Thirteen patients (31%) had mutations in nuclear genes not previously linked to disease. The pathogenicity of two such genes, NDUFB3 and AGK, was supported by complementation studies and evidence from multiple patients, respectively. The results underscore the potential and challenges of deploying NGS in clinical settings.
Conflict of interest statement
Competing interests: none.
Figures
Comment in
-
Disease genetics: Sequencing for diagnosis.Nat Rev Genet. 2012 Feb 7;13(3):150. doi: 10.1038/nrg3176. Nat Rev Genet. 2012. PMID: 22310895 No abstract available.
Similar articles
-
Targeted exome sequencing of suspected mitochondrial disorders.Neurology. 2013 May 7;80(19):1762-70. doi: 10.1212/WNL.0b013e3182918c40. Epub 2013 Apr 17. Neurology. 2013. PMID: 23596069 Free PMC article.
-
New genes and pathomechanisms in mitochondrial disorders unraveled by NGS technologies.Biochim Biophys Acta. 2016 Aug;1857(8):1326-1335. doi: 10.1016/j.bbabio.2016.02.022. Epub 2016 Mar 8. Biochim Biophys Acta. 2016. PMID: 26968897
-
New perspective in diagnostics of mitochondrial disorders: two years' experience with whole-exome sequencing at a national paediatric centre.J Transl Med. 2016 Jun 12;14(1):174. doi: 10.1186/s12967-016-0930-9. J Transl Med. 2016. PMID: 27290639 Free PMC article.
-
Diagnosis and molecular basis of mitochondrial respiratory chain disorders: exome sequencing for disease gene identification.Biochim Biophys Acta. 2014 Apr;1840(4):1355-9. doi: 10.1016/j.bbagen.2014.01.025. Epub 2014 Jan 24. Biochim Biophys Acta. 2014. PMID: 24462578 Review.
-
Next generation molecular diagnosis of mitochondrial disorders.Mitochondrion. 2013 Jul;13(4):379-87. doi: 10.1016/j.mito.2013.02.001. Epub 2013 Mar 6. Mitochondrion. 2013. PMID: 23473862 Review.
Cited by
-
The mitochondrial intermembrane space: the most constricted mitochondrial sub-compartment with the largest variety of protein import pathways.Open Biol. 2021 Mar;11(3):210002. doi: 10.1098/rsob.210002. Epub 2021 Mar 10. Open Biol. 2021. PMID: 33715390 Free PMC article. Review.
-
Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease.Ann Neurol. 2015 May;77(5):753-9. doi: 10.1002/ana.24362. Epub 2015 Mar 28. Ann Neurol. 2015. PMID: 25652200 Free PMC article.
-
A novel composition of two heterozygous GFM1 mutations in a Chinese child with epilepsy and mental retardation.Brain Behav. 2020 Oct;10(10):e01791. doi: 10.1002/brb3.1791. Epub 2020 Aug 9. Brain Behav. 2020. PMID: 32776492 Free PMC article.
-
Novel NDUFA13 Mutations Associated with OXPHOS Deficiency and Leigh Syndrome: A Second Family Report.Genes (Basel). 2020 Jul 26;11(8):855. doi: 10.3390/genes11080855. Genes (Basel). 2020. PMID: 32722639 Free PMC article.
-
Novel mutations in IBA57 are associated with leukodystrophy and variable clinical phenotypes.J Neurol. 2017 Jan;264(1):102-111. doi: 10.1007/s00415-016-8312-z. Epub 2016 Oct 26. J Neurol. 2017. PMID: 27785568
References
-
- Skladal D, Halliday J, Thorburn DR. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain. 2003;126:1905–12. - PubMed
-
- DiMauro S, Hirano M, Schon EA. Mitochondrial Medicine. Informa Healthcare; New York: 2006. p. 368.
-
- Dimauro S, Davidzon G. Mitochondrial DNA and disease. Ann Med. 2005;37:222–32. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
