

Whole-exome sequencing identifies homozygous AFG3L2 mutations in a spastic ataxia-neuropathy syndrome linked to mitochondrial m-AAA proteases
- PMID: 22022284
- PMCID: PMC3192828
- DOI: 10.1371/journal.pgen.1002325
Whole-exome sequencing identifies homozygous AFG3L2 mutations in a spastic ataxia-neuropathy syndrome linked to mitochondrial m-AAA proteases
Abstract
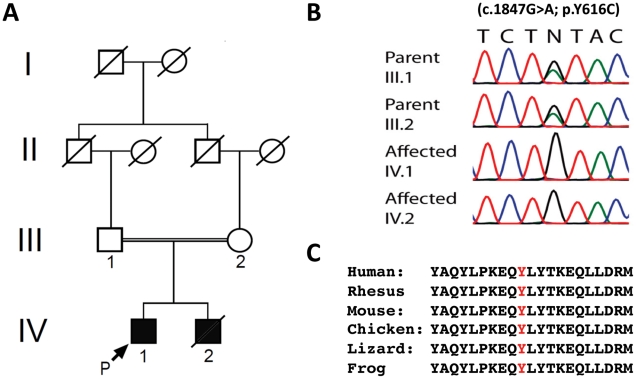
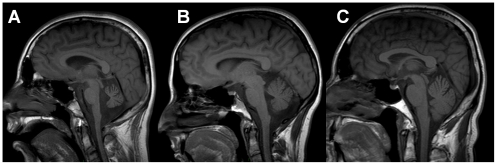
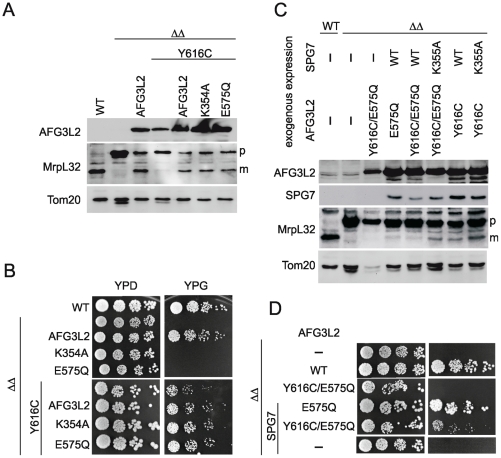
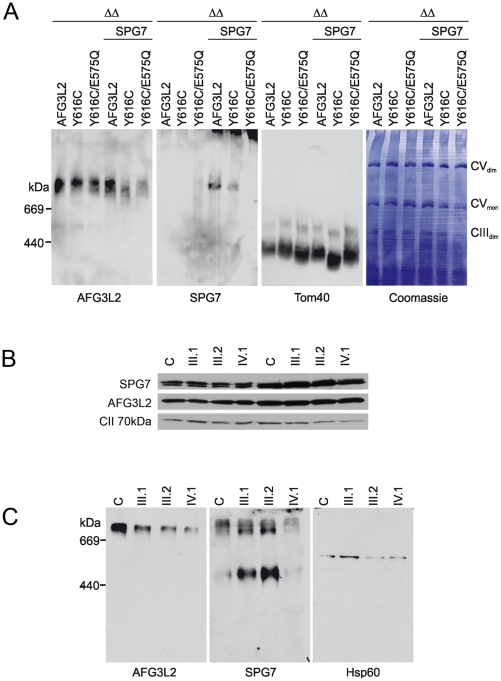
We report an early onset spastic ataxia-neuropathy syndrome in two brothers of a consanguineous family characterized clinically by lower extremity spasticity, peripheral neuropathy, ptosis, oculomotor apraxia, dystonia, cerebellar atrophy, and progressive myoclonic epilepsy. Whole-exome sequencing identified a homozygous missense mutation (c.1847G>A; p.Y616C) in AFG3L2, encoding a subunit of an m-AAA protease. m-AAA proteases reside in the mitochondrial inner membrane and are responsible for removal of damaged or misfolded proteins and proteolytic activation of essential mitochondrial proteins. AFG3L2 forms either a homo-oligomeric isoenzyme or a hetero-oligomeric complex with paraplegin, a homologous protein mutated in hereditary spastic paraplegia type 7 (SPG7). Heterozygous loss-of-function mutations in AFG3L2 cause autosomal-dominant spinocerebellar ataxia type 28 (SCA28), a disorder whose phenotype is strikingly different from that of our patients. As defined in yeast complementation assays, the AFG3L2(Y616C) gene product is a hypomorphic variant that exhibited oligomerization defects in yeast as well as in patient fibroblasts. Specifically, the formation of AFG3L2(Y616C) complexes was impaired, both with itself and to a greater extent with paraplegin. This produced an early-onset clinical syndrome that combines the severe phenotypes of SPG7 and SCA28, in additional to other "mitochondrial" features such as oculomotor apraxia, extrapyramidal dysfunction, and myoclonic epilepsy. These findings expand the phenotype associated with AFG3L2 mutations and suggest that AFG3L2-related disease should be considered in the differential diagnosis of spastic ataxias.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
m-AAA proteases, mitochondrial calcium homeostasis and neurodegeneration.Cell Res. 2018 Mar;28(3):296-306. doi: 10.1038/cr.2018.17. Epub 2018 Feb 16. Cell Res. 2018. PMID: 29451229 Free PMC article. Review.
-
Concurrent AFG3L2 and SPG7 mutations associated with syndromic parkinsonism and optic atrophy with aberrant OPA1 processing and mitochondrial network fragmentation.Hum Mutat. 2018 Dec;39(12):2060-2071. doi: 10.1002/humu.23658. Epub 2018 Oct 10. Hum Mutat. 2018. PMID: 30252181
-
Spinocerebellar Ataxia Type 28-Phenotypic and Molecular Characterization of a Family with Heterozygous and Compound-Heterozygous Mutations in AFG3L2.Cerebellum. 2019 Aug;18(4):817-822. doi: 10.1007/s12311-019-01036-2. Cerebellum. 2019. PMID: 31111429
-
Variable and tissue-specific subunit composition of mitochondrial m-AAA protease complexes linked to hereditary spastic paraplegia.Mol Cell Biol. 2007 Jan;27(2):758-67. doi: 10.1128/MCB.01470-06. Epub 2006 Nov 13. Mol Cell Biol. 2007. PMID: 17101804 Free PMC article.
-
Translating m-AAA protease function in mitochondria to hereditary spastic paraplegia.Trends Mol Med. 2006 Jun;12(6):262-9. doi: 10.1016/j.molmed.2006.04.002. Epub 2006 May 2. Trends Mol Med. 2006. PMID: 16647881 Review.
Cited by
-
Spastic paraplegia gene 7 in patients with spasticity and/or optic neuropathy.Brain. 2012 Oct;135(Pt 10):2980-93. doi: 10.1093/brain/aws240. Brain. 2012. PMID: 23065789 Free PMC article.
-
Mitochondrial ribosome assembly in health and disease.Cell Cycle. 2015;14(14):2226-50. doi: 10.1080/15384101.2015.1053672. Epub 2015 Jun 1. Cell Cycle. 2015. PMID: 26030272 Free PMC article. Review.
-
Genetic landscape remodelling in spinocerebellar ataxias: the influence of next-generation sequencing.J Neurol. 2015 Oct;262(10):2382-95. doi: 10.1007/s00415-015-7725-4. Epub 2015 Apr 11. J Neurol. 2015. PMID: 25862482 Review.
-
Mitochondrial quality control in the brain: The physiological and pathological roles.Front Neurosci. 2022 Dec 12;16:1075141. doi: 10.3389/fnins.2022.1075141. eCollection 2022. Front Neurosci. 2022. PMID: 36578825 Free PMC article. Review.
-
Mitochondrial HSP70 Chaperone System-The Influence of Post-Translational Modifications and Involvement in Human Diseases.Int J Mol Sci. 2021 Jul 28;22(15):8077. doi: 10.3390/ijms22158077. Int J Mol Sci. 2021. PMID: 34360841 Free PMC article. Review.
References
-
- Di Bella D, Lazzaro F, Brusco A, Plumari M, Battaglia G, et al. Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat Genet. 2010;42:313–321. - PubMed
-
- Cagnoli C, Stevanin G, Brussino A, Barberis M, Mancini C, et al. Missense mutations in the AFG3L2 proteolytic domain account for approximately 1.5% of European autosomal dominant cerebellar ataxias. Hum Mutat. 2010;31:1117–1124. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
