

Muscle choline kinase beta defect causes mitochondrial dysfunction and increased mitophagy
- PMID: 21750112
- PMCID: PMC3168292
- DOI: 10.1093/hmg/ddr305
Muscle choline kinase beta defect causes mitochondrial dysfunction and increased mitophagy
Abstract
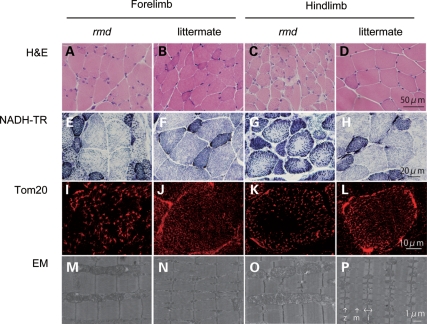
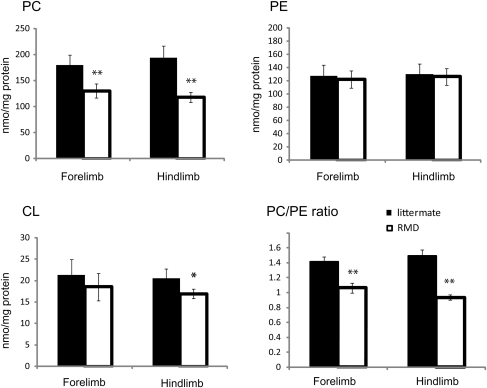
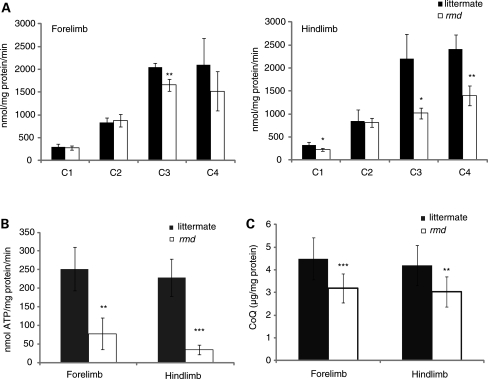
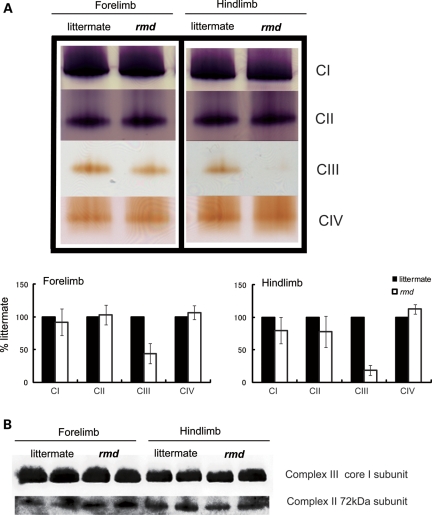
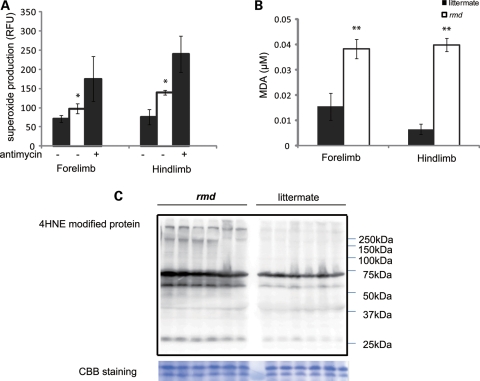
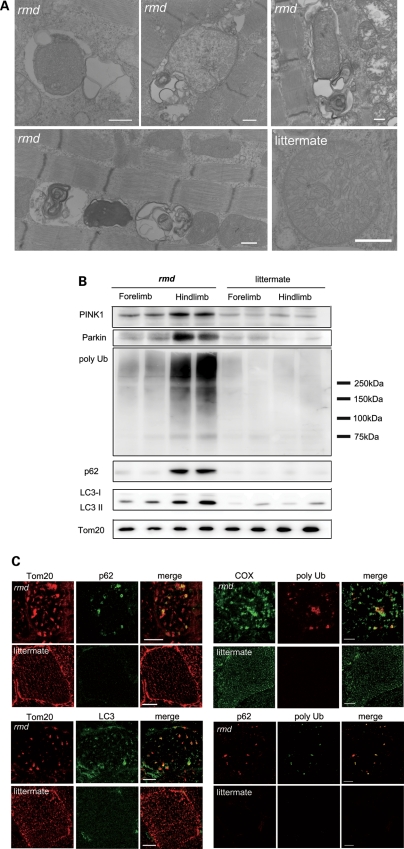
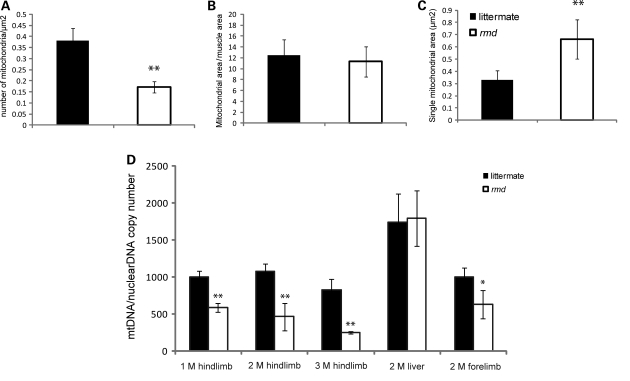
Choline kinase is the first step enzyme for phosphatidylcholine (PC) de novo biosynthesis. Loss of choline kinase activity in muscle causes rostrocaudal muscular dystrophy (rmd) in mouse and congenital muscular dystrophy in human, characterized by distinct mitochondrial morphological abnormalities. We performed biochemical and pathological analyses on skeletal muscle mitochondria from rmd mice. No mitochondria were found in the center of muscle fibers, while those located at the periphery of the fibers were significantly enlarged. Muscle mitochondria in rmd mice exhibited significantly decreased PC levels, impaired respiratory chain enzyme activities, decreased mitochondrial ATP synthesis, decreased coenzyme Q and increased superoxide production. Electron microscopy showed the selective autophagic elimination of mitochondria in rmd muscle. Molecular markers of mitophagy, including Parkin, PINK1, LC3, polyubiquitin and p62, were localized to mitochondria of rmd muscle. Quantitative analysis shows that the number of mitochondria in muscle fibers and mitochondrial DNA copy number were decreased. We demonstrated that the genetic defect in choline kinase in muscle results in mitochondrial dysfunction and subsequent mitochondrial loss through enhanced activation of mitophagy. These findings provide a first evidence for a pathomechanistic link between de novo PC biosynthesis and mitochondrial abnormality.
Figures
Similar articles
-
[New congenital muscular dystrophy due to CHKB mutations].Rinsho Shinkeigaku. 2013;53(11):1112-3. doi: 10.5692/clinicalneurol.53.1112. Rinsho Shinkeigaku. 2013. PMID: 24291895 Review. Japanese.
-
Megaconial congenital muscular dystrophy due to loss-of-function mutations in choline kinase β.Curr Opin Neurol. 2013 Oct;26(5):536-43. doi: 10.1097/WCO.0b013e328364c82d. Curr Opin Neurol. 2013. PMID: 23945283 Review.
-
A rostrocaudal muscular dystrophy caused by a defect in choline kinase beta, the first enzyme in phosphatidylcholine biosynthesis.J Biol Chem. 2006 Feb 24;281(8):4938-48. doi: 10.1074/jbc.M512578200. Epub 2005 Dec 21. J Biol Chem. 2006. PMID: 16371353
-
Phospholipid synthetic defect and mitophagy in muscle disease.Autophagy. 2011 Dec;7(12):1559-61. doi: 10.4161/auto.7.12.17925. Autophagy. 2011. PMID: 22024749 Free PMC article.
-
Functional rescue in a mouse model of congenital muscular dystrophy with megaconial myopathy.Hum Mol Genet. 2019 Aug 15;28(16):2635-2647. doi: 10.1093/hmg/ddz068. Hum Mol Genet. 2019. PMID: 31216357 Free PMC article.
Cited by
-
Lipid-induced mitochondrial stress and insulin action in muscle.Cell Metab. 2012 May 2;15(5):595-605. doi: 10.1016/j.cmet.2012.04.010. Cell Metab. 2012. PMID: 22560212 Free PMC article.
-
Tricarboxylic acid (TCA) cycle, sphingolipid, and phosphatidylcholine metabolism are dysregulated in T. gondii infection-induced cachexia.Heliyon. 2023 Jul 5;9(7):e17411. doi: 10.1016/j.heliyon.2023.e17411. eCollection 2023 Jul. Heliyon. 2023. PMID: 37456044 Free PMC article.
-
Divergent Skeletal Muscle Metabolomic Signatures of Different Exercise Training Modes Independently Predict Cardiometabolic Risk Factors.Diabetes. 2024 Jan 1;73(1):23-37. doi: 10.2337/db23-0142. Diabetes. 2024. PMID: 37862464 Free PMC article.
-
Phospholipids: Identification and Implication in Muscle Pathophysiology.Int J Mol Sci. 2021 Jul 30;22(15):8176. doi: 10.3390/ijms22158176. Int J Mol Sci. 2021. PMID: 34360941 Free PMC article. Review.
-
Megaconial congenital muscular dystrophy due to novel CHKB variants: a case report and literature review.Skelet Muscle. 2022 Sep 29;12(1):23. doi: 10.1186/s13395-022-00306-8. Skelet Muscle. 2022. PMID: 36175989 Free PMC article. Review.
References
-
- Mitsuhashi S., Ohkuma A., Talim B., Karahashi M., Koumura T., Aoyama C., Kurihara M., Quinlivan R., Sewry C., Mitsuhashi H., et al. A congenital muscular dystrophy with mitochondrial structural abnormalities caused by defective de novo phosphatidylcholine biosynthesis. Am. J. Hum. Genet. 2011;88:845–851. doi:10.1016/j.ajhg.2011.05.010. - DOI - PMC - PubMed
-
- Sher R.B., Aoyama C., Huebsch K.A., Ji S., Kerner J., Yang Y., Frankel W.A., Hoppel C.A., Wood P.A., Vance D.E., et al. A rostrocaudal muscular dystrophy caused by a defect in choline kinase beta, the first enzyme in phosphatidylcholine biosynthesis. J. Biol. Chem. 2006;281:4938–4948. doi:10.1074/jbc.M512578200. - DOI - PubMed
-
- Saraste M. Oxidative phosphorylation at the fin de siècle. Science. 1999;283:1488–1493. doi:10.1126/science.283.5407.1488. - DOI - PubMed
-
- Spierings D., McStay G., Saleh M., Bender C., Chipuk J., Maurer U., Green D.R. Connected to death: the (unexpurgated) mitochondrial pathway of apoptosis. Science. 2005;310:66–67. doi:10.1126/science.1117105. - DOI - PubMed
-
- Wakabayashi T. Megamitochondria formation—physiology and pathology. J. Cell Mol. Med. 2002;6:497–538. doi:10.1111/j.1582-4934.2002.tb00452.x. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
