

Guidelines for the diagnosis and management of chylomicron retention disease based on a review of the literature and the experience of two centers
- PMID: 20920215
- PMCID: PMC2956717
- DOI: 10.1186/1750-1172-5-24
Guidelines for the diagnosis and management of chylomicron retention disease based on a review of the literature and the experience of two centers
Abstract
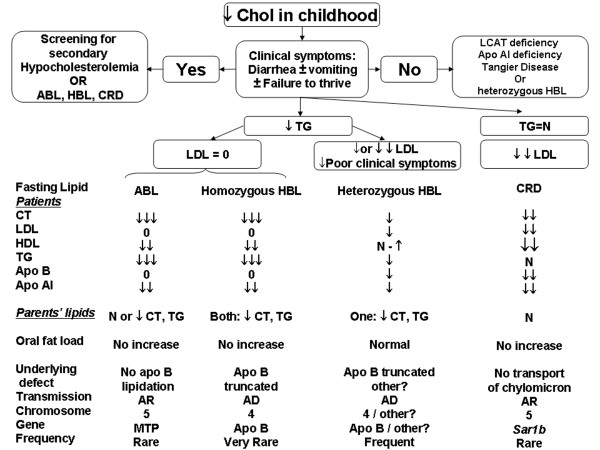
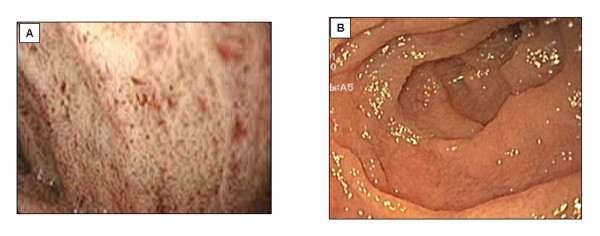
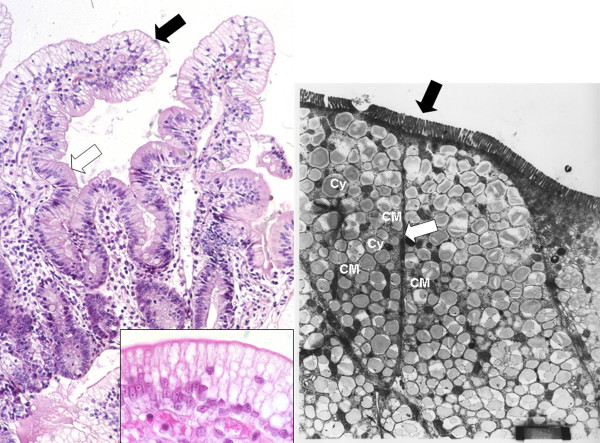
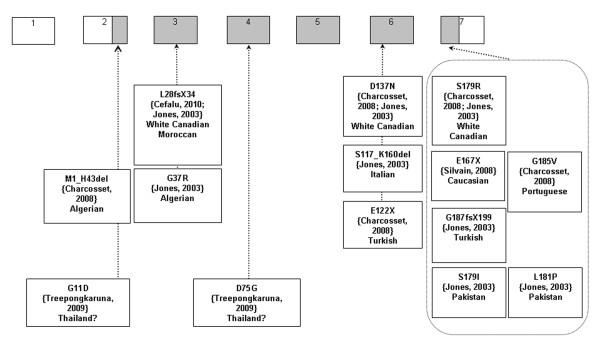
Familial hypocholesterolemia, namely abetalipoproteinemia, hypobetalipoproteinemia and chylomicron retention disease (CRD), are rare genetic diseases that cause malnutrition, failure to thrive, growth failure and vitamin E deficiency, as well as other complications. Recently, the gene implicated in CRD was identified. The diagnosis is often delayed because symptoms are nonspecific. Treatment and follow-up remain poorly defined.The aim of this paper is to provide guidelines for the diagnosis, treatment and follow-up of children with CRD based on a literature overview and two pediatric centers 'experience.The diagnosis is based on a history of chronic diarrhea with fat malabsorption and abnormal lipid profile. Upper endoscopy and histology reveal fat-laden enterocytes whereas vitamin E deficiency is invariably present. Creatine kinase (CK) is usually elevated and hepatic steatosis is common. Genotyping identifies the Sar1b gene mutation.Treatment should be aimed at preventing potential complications. Vomiting, diarrhea and abdominal distension improve on a low-long chain fat diet. Failure to thrive is one of the most common initial clinical findings. Neurological and ophthalmologic complications in CRD are less severe than in other types of familial hypocholesterolemia. However, the vitamin E deficiency status plays a pivotal role in preventing neurological complications. Essential fatty acid (EFA) deficiency is especially severe early in life. Recently, increased CK levels and cardiomyopathy have been described in addition to muscular manifestations. Poor mineralization and delayed bone maturation do occur. A moderate degree of macrovesicular steatosis is common, but no cases of steatohepatitis cirrhosis. Besides a low-long chain fat diet made up uniquely of polyunsaturated fatty acids, treatment includes fat-soluble vitamin supplements and large amounts of vitamin E. Despite fat malabsorption and the absence of postprandial chylomicrons, the oral route can prevent neurological complications even though serum levels of vitamin E remain chronically low. Dietary counseling is needed not only to monitor fat intake and improve symptoms, but also to maintain sufficient caloric and EFA intake. Despite a better understanding of the pathogenesis of CRD, the diagnosis and management of the disease remain a challenge for clinicians. The clinical guidelines proposed will helpfully lead to an earlier diagnosis and the prevention of complications.
Figures
Similar articles
-
Chylomicron retention disease: a long term study of two cohorts.Mol Genet Metab. 2009 Jun;97(2):136-42. doi: 10.1016/j.ymgme.2009.02.003. Epub 2009 Feb 20. Mol Genet Metab. 2009. PMID: 19285442
-
Chylomicron retention disease: report of two cases from a Greek Island.J Pediatr Endocrinol Metab. 2012;25(11-12):1191-4. doi: 10.1515/jpem-2012-0243. J Pediatr Endocrinol Metab. 2012. PMID: 23329770
-
[Chylomicron retention disease caused by SAR1B gene variations in 2 cases and literatures review].Zhonghua Er Ke Za Zhi. 2024 Jun 2;62(6):565-570. doi: 10.3760/cma.j.cn112140-20240301-00138. Zhonghua Er Ke Za Zhi. 2024. PMID: 38763880 Review. Chinese.
-
Chylomicron retention disease caused by a new pathogenic variant in sar1b protein: a rare case report from Syria.BMC Pediatr. 2021 Oct 11;21(1):449. doi: 10.1186/s12887-021-02897-5. BMC Pediatr. 2021. PMID: 34629076 Free PMC article.
-
Chylomicron retention disease: genetics, biochemistry, and clinical spectrum.Curr Opin Lipidol. 2019 Apr;30(2):134-139. doi: 10.1097/MOL.0000000000000578. Curr Opin Lipidol. 2019. PMID: 30640893 Review.
Cited by
-
Validation of Knock-Out Caco-2 TC7 Cells as Models of Enterocytes of Patients with Familial Genetic Hypobetalipoproteinemias.Nutrients. 2023 Jan 18;15(3):505. doi: 10.3390/nu15030505. Nutrients. 2023. PMID: 36771214 Free PMC article.
-
Extremely low levels of low-density lipoprotein potentially suggestive of familial hypobetalipoproteinemia: A separate phenotype of NAFLD?J Clin Lipidol. 2019 May-Jun;13(3):425-431. doi: 10.1016/j.jacl.2019.02.002. Epub 2019 Feb 14. J Clin Lipidol. 2019. PMID: 30879942 Free PMC article.
-
Current Diagnosis and Management of Abetalipoproteinemia.J Atheroscler Thromb. 2021 Oct 1;28(10):1009-1019. doi: 10.5551/jat.RV17056. Epub 2021 May 16. J Atheroscler Thromb. 2021. PMID: 33994405 Free PMC article. Review.
-
Plasma and tissue concentrations of α-tocopherol and δ-tocopherol following high dose dietary supplementation in mice.Nutrients. 2012 Jun;4(6):467-90. doi: 10.3390/nu4060467. Epub 2012 Jun 6. Nutrients. 2012. PMID: 22822447 Free PMC article.
-
SAR1B GTPase is necessary to protect intestinal cells from disorders of lipid homeostasis, oxidative stress, and inflammation.J Lipid Res. 2019 Oct;60(10):1755-1764. doi: 10.1194/jlr.RA119000119. Epub 2019 Aug 13. J Lipid Res. 2019. PMID: 31409740 Free PMC article.
References
-
- Leiper JM, Bayliss JD, Pease RJ, Brett DJ, Scott J, Shoulders CC. Microsomal triglyceride transfer protein, the abetalipoproteinemia gene product, mediates the secretion of apolipoprotein B-containing lipoproteins from heterologous cells. J Biol Chem. 1994;269:21951–21954. - PubMed
-
- Noto D, Cefalu AB, Cannizzaro A, Mina M, Fayer F, Valenti V, Barbagallo CM, Tuttolomondo A, Pinto A, Sciume C, Licata G, Averna M. Familial hypobetalipoproteinemia due to apolipoprotein B R463W mutation causes intestinal fat accumulation and low postprandial lipemia. Atherosclerosis. 2009;206:193–198. doi: 10.1016/j.atherosclerosis.2009.01.037. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
