

Defective FA2H leads to a novel form of neurodegeneration with brain iron accumulation (NBIA)
- PMID: 20853438
- PMCID: PMC6059612
- DOI: 10.1002/ana.22122
Defective FA2H leads to a novel form of neurodegeneration with brain iron accumulation (NBIA)
Abstract
Objective: Neurodegeneration with brain iron accumulation (NBIA) represents a distinctive phenotype of neurodegenerative disease for which several causative genes have been identified. The spectrum of neurologic disease associated with mutations in NBIA genes is broad, with phenotypes that range from infantile neurodegeneration and death in childhood to adult-onset parkinsonism-dystonia. Here we report the discovery of a novel gene that leads to a distinct form of NBIA.
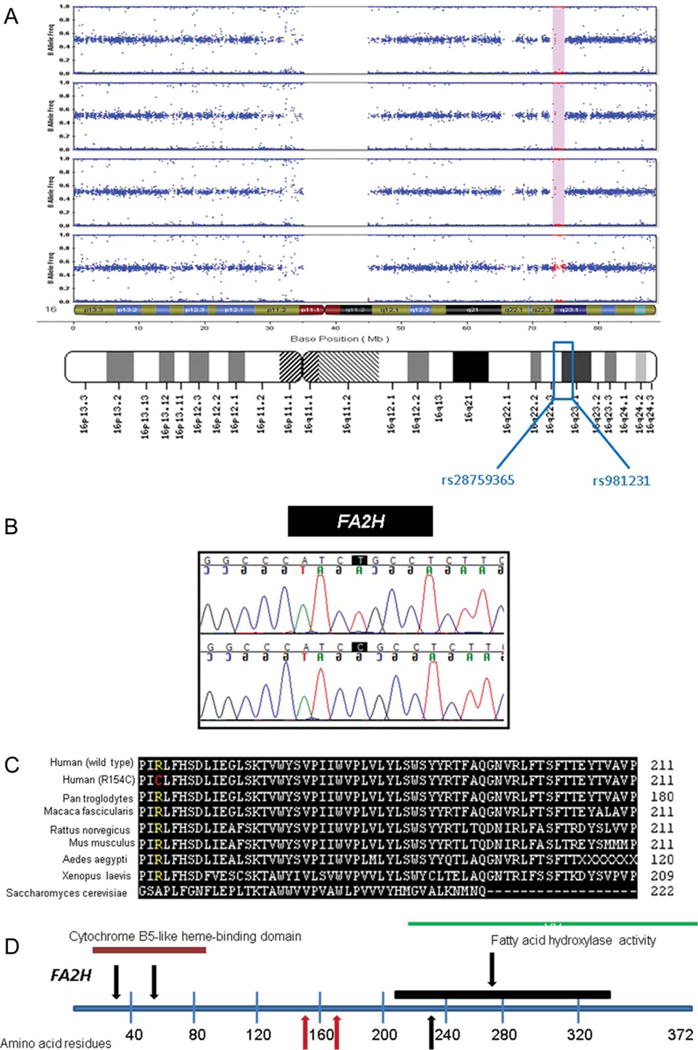
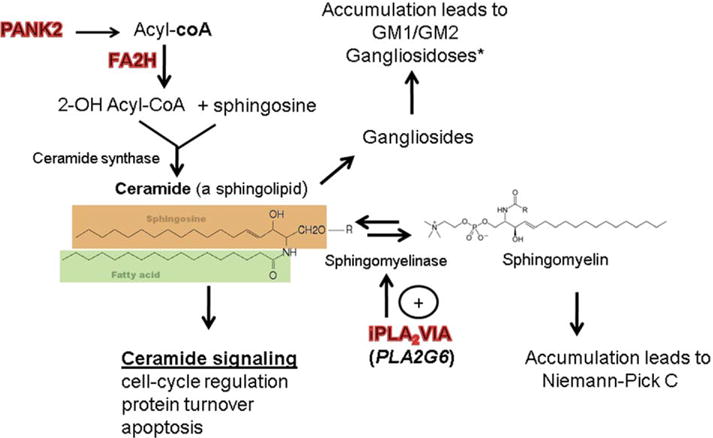
Methods: Using autozygosity mapping and candidate gene sequencing, we identified mutations in the fatty acid hydroxylase gene FA2H, newly implicating abnormalities of ceramide metabolism in the pathogenesis of NBIA.
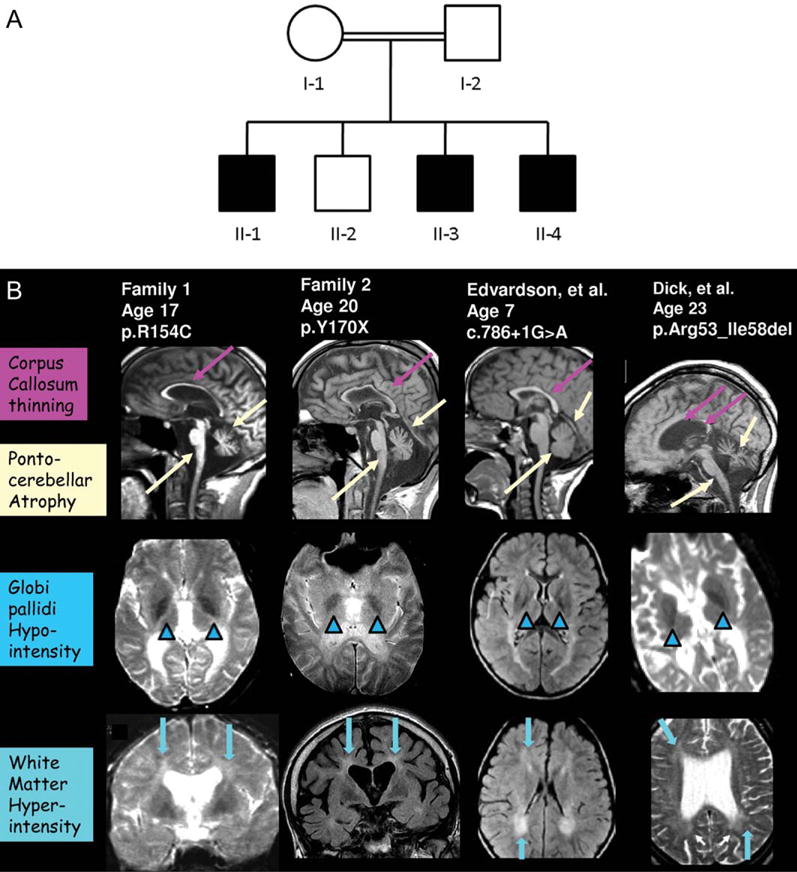
Results: Neuroimaging demonstrated T2 hypointensity in the globus pallidus, confluent T2 white matter hyperintensities, and profound pontocerebellar atrophy in affected members of two families. Phenotypically, affected family members exhibited spastic quadriparesis, ataxia, and dystonia with onset in childhood and episodic neurological decline. Analogous to what has been reported previously for PLA2G6, the phenotypic spectrum of FA2H mutations is diverse based on our findings and those of prior investigators, because FA2H mutations have been identified in both a form of hereditary spastic paraplegia (SPG35) and a progressive familial leukodystrophy.
Interpretation: These findings link white matter degeneration and NBIA for the first time and implicate new signaling pathways in the genesis of NBIA.
Conflict of interest statement
Nothing to report.
Figures
Comment in
-
Three faces of the same gene: FA2H links neurodegeneration with brain iron accumulation, leukodystrophies, and hereditary spastic paraplegias.Ann Neurol. 2010 Nov;68(5):575-7. doi: 10.1002/ana.22211. Ann Neurol. 2010. PMID: 21031573 No abstract available.
Similar articles
-
FAHN/SPG35: a narrow phenotypic spectrum across disease classifications.Brain. 2019 Jun 1;142(6):1561-1572. doi: 10.1093/brain/awz102. Brain. 2019. PMID: 31135052 Free PMC article.
-
Hereditary spastic paraplegia type 35 in a Turkish girl with fatty acid hydroxylase-associated neurodegeneration.J Pediatr Endocrinol Metab. 2024 Feb 6;37(3):271-275. doi: 10.1515/jpem-2023-0481. Print 2024 Mar 25. J Pediatr Endocrinol Metab. 2024. PMID: 38353247
-
Novel biallelic FA2H mutations in a Japanese boy with fatty acid hydroxylase-associated neurodegeneration.Brain Dev. 2020 Feb;42(2):217-221. doi: 10.1016/j.braindev.2019.11.006. Epub 2019 Dec 16. Brain Dev. 2020. PMID: 31837835
-
[GENETICALLY DETERMINED DISEASES ASSOCIATED WITH PATHOLOGICAL BRAIN IRON ACCUMULATION AND NEURODEGENERATION].Ideggyogy Sz. 2016 Mar 30;69(5-6):157-66. doi: 10.18071/isz.69.0157. Ideggyogy Sz. 2016. PMID: 27468605 Review. Hungarian.
-
Clinical and neuroimaging features of autosomal recessive spastic paraplegia 35 (SPG35): case reports, new mutations, and brief literature review.Neurogenetics. 2018 May;19(2):123-130. doi: 10.1007/s10048-018-0538-8. Epub 2018 Feb 8. Neurogenetics. 2018. PMID: 29423566 Review.
Cited by
-
Review: Insights into molecular mechanisms of disease in neurodegeneration with brain iron accumulation: unifying theories.Neuropathol Appl Neurobiol. 2016 Apr;42(3):220-41. doi: 10.1111/nan.12242. Epub 2015 Jun 2. Neuropathol Appl Neurobiol. 2016. PMID: 25870938 Free PMC article. Review.
-
Pathophysiology and treatment of neurodegeneration with brain iron accumulation in the pediatric population.Curr Treat Options Neurol. 2013 Oct;15(5):652-67. doi: 10.1007/s11940-013-0254-5. Curr Treat Options Neurol. 2013. PMID: 23888388
-
Clinical approach to Parkinson's disease: features, diagnosis, and principles of management.Cold Spring Harb Perspect Med. 2012 Jun;2(6):a008870. doi: 10.1101/cshperspect.a008870. Cold Spring Harb Perspect Med. 2012. PMID: 22675666 Free PMC article.
-
Myelin lipid metabolism and its role in myelination and myelin maintenance.Innovation (Camb). 2022 Dec 7;4(1):100360. doi: 10.1016/j.xinn.2022.100360. eCollection 2023 Jan 30. Innovation (Camb). 2022. PMID: 36588745 Free PMC article. Review.
-
Fatty Acid 2-Hydroxylase and 2-Hydroxylated Sphingolipids: Metabolism and Function in Health and Diseases.Int J Mol Sci. 2023 Mar 3;24(5):4908. doi: 10.3390/ijms24054908. Int J Mol Sci. 2023. PMID: 36902339 Free PMC article. Review.
References
-
- Thomas M, Hayflick SJ, Jankovic J. Clinical heterogeneity of neurodegeneration with brain iron accumulation (Hallervorden-Spatz syndrome) and pantothenate kinase-associated neurodegeneration. Mov Disord. 2004;19:36–42. - PubMed
-
- Curtis ARJ, Fey C, Morris CM, et al. Mutation in the gene encoding ferritin light polypeptide causes dominant adult-onset basal ganglia disease. Nat Genet. 2001;28:350–354. - PubMed
-
- Zhou B, Westaway SK, Levinson B, et al. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat Genet. 2001;28:345–349. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
