

Succinyl-CoA ligase deficiency: a mitochondrial hepatoencephalomyopathy
- PMID: 20453710
- PMCID: PMC2928220
- DOI: 10.1203/PDR.0b013e3181e5c3a4
Succinyl-CoA ligase deficiency: a mitochondrial hepatoencephalomyopathy
Abstract
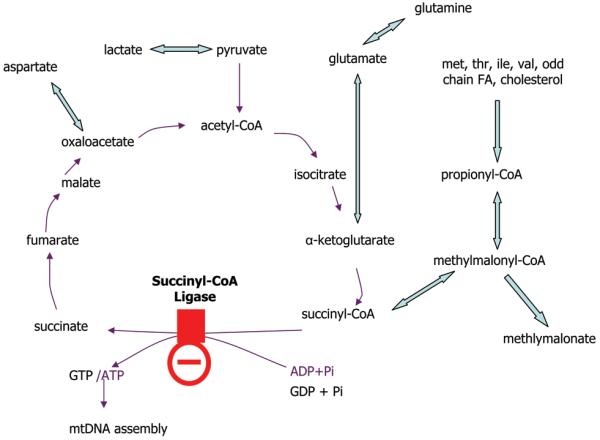
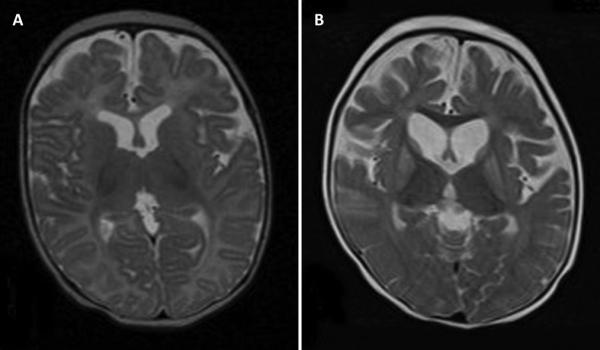
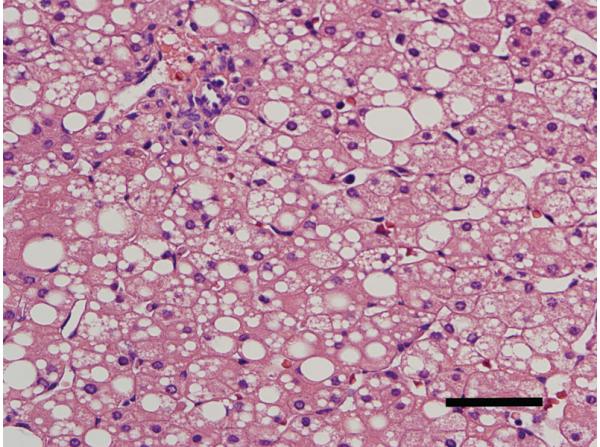
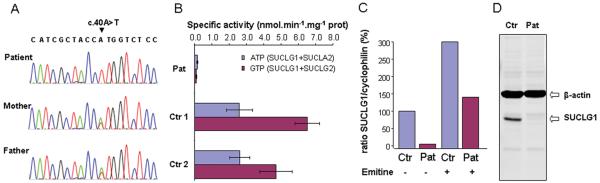
This patient presented on the first day of life with pronounced lactic acidosis with an elevated lactate/pyruvate ratio. Urine organic acids showed Krebs cycle metabolites and mildly elevated methylmalonate and methylcitrate. The acylcarnitine profile showed elevated propionylcarnitine and succinylcarnitine. Amino acids showed elevated glutamic acid, glutamine, proline, and alanine. From the age 2 of mo on, she had elevated transaminases and intermittent episodes of liver failure. Liver biopsy showed steatosis and a decrease of mitochondrial DNA to 50% of control. She had bilateral sensorineural hearing loss. Over the course of the first 2 y of life, she developed a progressively severe myopathy with pronounced muscle weakness eventually leading to respiratory failure, Leigh disease, and recurrent hepatic failure. The hepatic symptoms and the metabolic parameters temporarily improved on treatment with aspartate, but neither muscle symptoms nor brain lesions improved. Laboratory testing revealed a deficiency of succinyl-CoA ligase enzyme activity and protein in fibroblasts because of a novel homozygous mutation in the SUCLG1 gene: c.40A>T (p.M14L). Functional analysis suggests that this methionine is more likely to function as the translation initiator methionine, explaining the pathogenic nature of the mutation. Succinyl-CoA ligase deficiency due to an SUCLG1 mutation is a new cause for mitochondrial hepatoencephalomyopathy.
Figures

Similar articles
-
Succinyl-CoA synthetase (SUCLA2) deficiency in two siblings with impaired activity of other mitochondrial oxidative enzymes in skeletal muscle without mitochondrial DNA depletion.Mol Genet Metab. 2017 Mar;120(3):213-222. doi: 10.1016/j.ymgme.2016.11.005. Epub 2016 Nov 12. Mol Genet Metab. 2017. PMID: 27913098 Free PMC article.
-
Five novel SUCLG1 mutations in three Chinese patients with succinate-CoA ligase deficiency noticed by mild methylmalonic aciduria.Brain Dev. 2016 Jan;38(1):61-7. doi: 10.1016/j.braindev.2015.05.002. Epub 2015 May 28. Brain Dev. 2016. PMID: 26028457
-
Succinate-CoA ligase deficiency due to mutations in SUCLA2 and SUCLG1: phenotype and genotype correlations in 71 patients.J Inherit Metab Dis. 2016 Mar;39(2):243-52. doi: 10.1007/s10545-015-9894-9. Epub 2015 Oct 16. J Inherit Metab Dis. 2016. PMID: 26475597
-
Disorders caused by deficiency of succinate-CoA ligase.J Inherit Metab Dis. 2008 Apr;31(2):226-9. doi: 10.1007/s10545-008-0828-7. Epub 2008 Apr 4. J Inherit Metab Dis. 2008. PMID: 18392745 Review.
-
Infantile mitochondrial disorders.Biosci Rep. 2007 Jun;27(1-3):105-12. doi: 10.1007/s10540-007-9039-y. Biosci Rep. 2007. PMID: 17486440 Review.
Cited by
-
Liver X receptors in lipid metabolism: opportunities for drug discovery.Nat Rev Drug Discov. 2014 Jun;13(6):433-44. doi: 10.1038/nrd4280. Epub 2014 May 16. Nat Rev Drug Discov. 2014. PMID: 24833295 Review.
-
Beyond mitochondria: Alternative energy-producing pathways from all strata of life.Metabolism. 2021 May;118:154733. doi: 10.1016/j.metabol.2021.154733. Epub 2021 Feb 23. Metabolism. 2021. PMID: 33631145 Free PMC article. Review.
-
Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation.Biochem J. 2011 Feb 1;433(3):505-14. doi: 10.1042/BJ20100791. Biochem J. 2011. PMID: 21044047 Free PMC article.
-
Mitochondrial DNA copy number in human disease: the more the better?FEBS Lett. 2021 Apr;595(8):976-1002. doi: 10.1002/1873-3468.14021. Epub 2020 Dec 25. FEBS Lett. 2021. PMID: 33314045 Free PMC article. Review.
-
Novel compound heterozygous SUCLG1 variants may contribute to mitochondria DNA depletion syndrome-9.Mol Genet Genomic Med. 2022 Sep;10(9):e2010. doi: 10.1002/mgg3.2010. Epub 2022 Jun 28. Mol Genet Genomic Med. 2022. PMID: 35762302 Free PMC article.
References
-
- Alberio S, Mineri R, Tiranti V, Zeviani M. Depletion of mtDNA: syndromes and genes. Mitochondrion. 2007;7:6–12. - PubMed
-
- Johnson JD, Muhonen WW, Lambeth DO. Characterization of the ATP- and GTP-specific succinyl-CoA synthetases in pigeon. J Biol Chem. 1998;273:27573–27579. - PubMed
-
- Johnson JD, Mehus JG, Tews K, Milavetz BL, Lambeth DO. Genetic evidence for the expression of ATP- and GTP-specific succinyl-CoA synthetases in multicellular eucaryotes. J Biol Chem. 1998;273:27580–27586. - PubMed
-
- Lambeth DO, Tews KN, Adkins S, Frohlich D, Milavetz BL. Expression of two succinyl-CoA synthetases with different nucleotide specificities in mammalian tissues. J Biol Chem. 2004;279:36621–36624. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
