

A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay
- PMID: 20154674
- PMCID: PMC2847896
- DOI: 10.1038/ng.534
A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay
Abstract
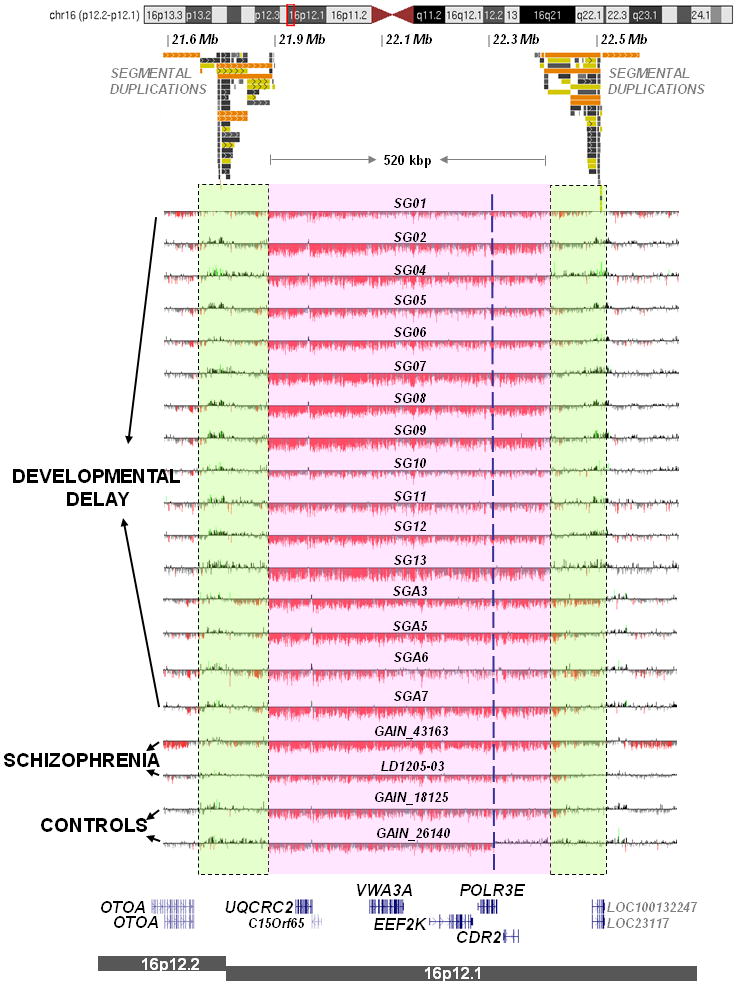
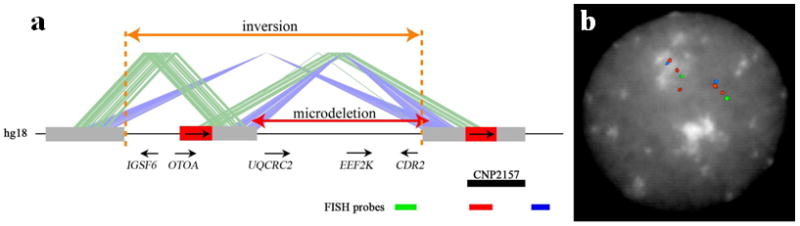
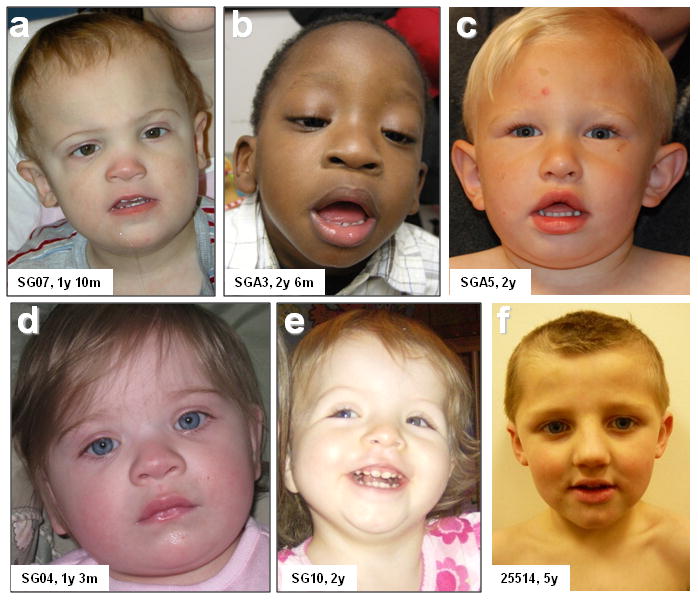
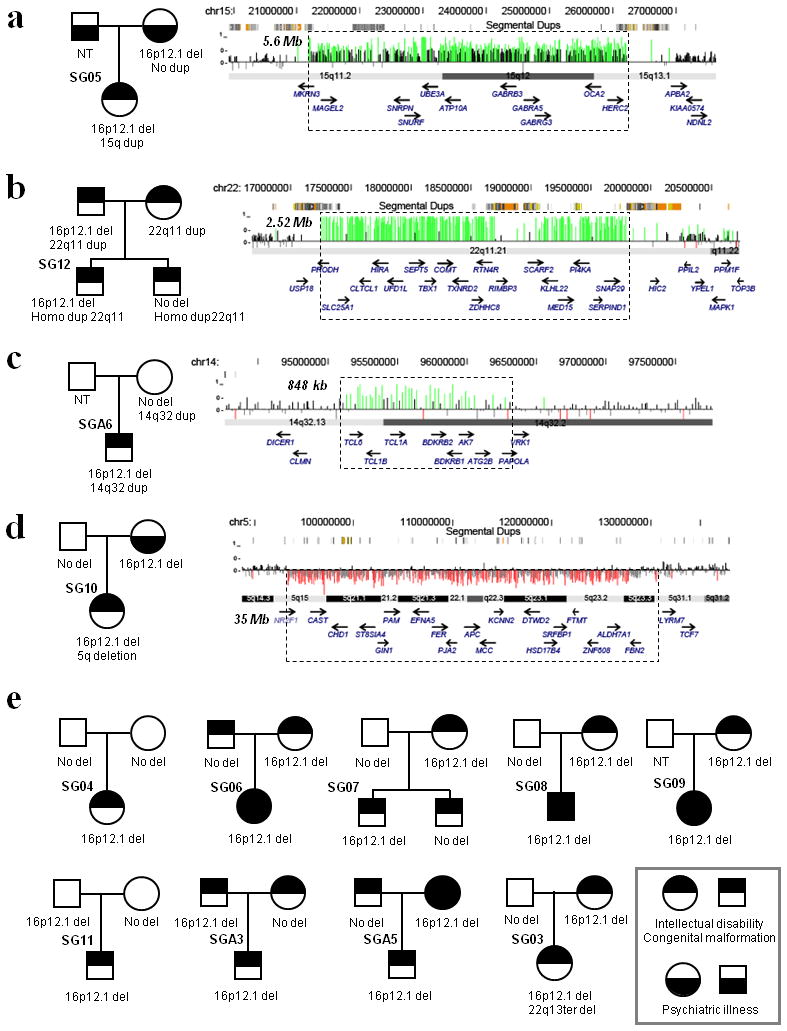
We report the identification of a recurrent, 520-kb 16p12.1 microdeletion associated with childhood developmental delay. The microdeletion was detected in 20 of 11,873 cases compared with 2 of 8,540 controls (P = 0.0009, OR = 7.2) and replicated in a second series of 22 of 9,254 cases compared with 6 of 6,299 controls (P = 0.028, OR = 2.5). Most deletions were inherited, with carrier parents likely to manifest neuropsychiatric phenotypes compared to non-carrier parents (P = 0.037, OR = 6). Probands were more likely to carry an additional large copy-number variant when compared to matched controls (10 of 42 cases, P = 5.7 x 10(-5), OR = 6.6). The clinical features of individuals with two mutations were distinct from and/or more severe than those of individuals carrying only the co-occurring mutation. Our data support a two-hit model in which the 16p12.1 microdeletion both predisposes to neuropsychiatric phenotypes as a single event and exacerbates neurodevelopmental phenotypes in association with other large deletions or duplications. Analysis of other microdeletions with variable expressivity indicates that this two-hit model might be more generally applicable to neuropsychiatric disease.
Conflict of interest statement
Figures
Comment in
-
Understanding variable expressivity in microdeletion syndromes.Nat Genet. 2010 Mar;42(3):192-3. doi: 10.1038/ng0310-192. Nat Genet. 2010. PMID: 20179732 No abstract available.
-
Journal club. A neurodevelopmental geneticist explores how one mutation can lead to multiple diseases.Nature. 2010 Apr 22;464(7292):1107. doi: 10.1038/4641107e. Nature. 2010. PMID: 20414273 No abstract available.
-
Two-hit wonder: a novel genetic model to explain variable expressivity in severe pediatric phenotypes.Clin Genet. 2010 Dec;78(6):517-9. doi: 10.1111/j.1399-0004.2010.01530_1.x. Epub 2010 Sep 29. Clin Genet. 2010. PMID: 20880121 No abstract available.
Similar articles
-
Refining the Phenotype of Recurrent Rearrangements of Chromosome 16.Int J Mol Sci. 2019 Mar 4;20(5):1095. doi: 10.3390/ijms20051095. Int J Mol Sci. 2019. PMID: 30836598 Free PMC article.
-
Cervicomedullary spinal stenosis and ventriculomegaly in a child with developmental delay due to chromosome 16p12.1 microdeletion syndrome.J Child Neurol. 2015 Mar;30(3):394-6. doi: 10.1177/0883073814533149. Epub 2014 May 9. J Child Neurol. 2015. PMID: 24813870
-
Combinatorial patterns of gene expression changes contribute to variable expressivity of the developmental delay-associated 16p12.1 deletion.Genome Med. 2021 Oct 18;13(1):163. doi: 10.1186/s13073-021-00982-z. Genome Med. 2021. PMID: 34657631 Free PMC article.
-
An additional case of the recurrent 15q24.1 microdeletion syndrome and review of the literature.Twin Res Hum Genet. 2011 Aug;14(4):333-9. doi: 10.1375/twin.14.4.333. Twin Res Hum Genet. 2011. PMID: 21787116 Review.
-
The discovery of microdeletion syndromes in the post-genomic era: review of the methodology and characterization of a new 1q41q42 microdeletion syndrome.Genet Med. 2007 Sep;9(9):607-16. doi: 10.1097/gim.0b013e3181484b49. Genet Med. 2007. PMID: 17873649 Review.
Cited by
-
Sixteen New Cases Contributing to the Characterization of Patients with Distal 22q11.2 Microduplications.Mol Syndromol. 2010;1(5):246-254. doi: 10.1159/000327982. Epub 2011 May 18. Mol Syndromol. 2010. PMID: 22140377 Free PMC article.
-
What is complex about complex disorders?Genome Biol. 2012 Jan 23;13(1):237. doi: 10.1186/gb-2012-13-1-237. Genome Biol. 2012. PMID: 22269335 Free PMC article.
-
Novel candidate genes and regions for childhood apraxia of speech identified by array comparative genomic hybridization.Genet Med. 2012 Nov;14(11):928-36. doi: 10.1038/gim.2012.72. Epub 2012 Jul 5. Genet Med. 2012. PMID: 22766611 Free PMC article.
-
Array-Based Comparative Genomic Hybridization Analysis in Children with Developmental Delay/Intellectual Disability.Balkan J Med Genet. 2022 Jun 5;24(2):15-24. doi: 10.2478/bjmg-2021-0020. eCollection 2021 Nov. Balkan J Med Genet. 2022. PMID: 36249514 Free PMC article.
-
Chromosomal microarray analysis in clinical evaluation of neurodevelopmental disorders-reporting a novel deletion of SETDB1 and illustration of counseling challenge.Pediatr Res. 2016 Sep;80(3):371-81. doi: 10.1038/pr.2016.101. Epub 2016 Apr 27. Pediatr Res. 2016. PMID: 27119313 Free PMC article.
References
-
- Lupski JR. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–22. - PubMed
-
- Eichler EE, et al. Divergent origins and concerted expansion of two segmental duplications on chromosome 16. J Hered. 2001;92:462–8. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
