

FG syndrome, an X-linked multiple congenital anomaly syndrome: the clinical phenotype and an algorithm for diagnostic testing
- PMID: 19938245
- PMCID: PMC4113033
- DOI: 10.1097/GIM.0b013e3181bd3d90
FG syndrome, an X-linked multiple congenital anomaly syndrome: the clinical phenotype and an algorithm for diagnostic testing
Abstract
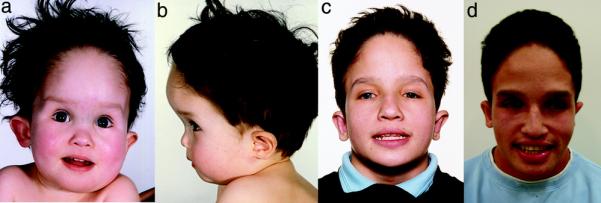
FG syndrome is a rare X-linked multiple congenital anomaly-cognitive impairment disorder caused by the p.R961W mutation in the MED12 gene. We identified all known patients with this mutation to delineate their clinical phenotype and devise a clinical algorithm to facilitate molecular diagnosis. We ascertained 23 males with the p.R961W mutation in MED12 from 9 previously reported FG syndrome families and 1 new family. Six patients are reviewed in detail. These 23 patients were compared with 48 MED12 mutation-negative patients, who had the clinical diagnosis of FG syndrome. Traits that best discriminated between these two groups were chosen to develop an algorithm with high sensitivity and specificity for the p.R961W MED12 mutation. FG syndrome has a recognizable dysmorphic phenotype with a high incidence of congenital anomalies. A family history of X-linked mental retardation, deceased male infants, and/or multiple fetal losses was documented in all families. The algorithm identifies the p.R961W MED12 mutation-positive group with 100% sensitivity and 90% specificity. The clinical phenotype of FG syndrome defines a recognizable pattern of X-linked multiple congenital anomalies and cognitive impairment. This algorithm can assist the clinician in selecting the patients for testing who are most likely to have the recurrent p.R961W MED12 mutation.
Figures

Similar articles
-
Behavior of 10 patients with FG syndrome (Opitz-Kaveggia syndrome) and the p.R961W mutation in the MED12 gene.Am J Med Genet A. 2008 Dec 1;146A(23):3011-7. doi: 10.1002/ajmg.a.32553. Am J Med Genet A. 2008. PMID: 18973276 Free PMC article.
-
Clinical experience in the evaluation of 30 patients with a prior diagnosis of FG syndrome.J Med Genet. 2009 Jan;46(1):9-13. doi: 10.1136/jmg.2008.060509. Epub 2008 Sep 19. J Med Genet. 2009. PMID: 18805826
-
A novel mutation in MED12 causes FG syndrome (Opitz-Kaveggia syndrome).Clin Genet. 2011 Feb;79(2):183-8. doi: 10.1111/j.1399-0004.2010.01449.x. Clin Genet. 2011. PMID: 20507344
-
MED12 missense mutation in a three-generation family. Clinical characterization of MED12-related disorders and literature review.Eur J Med Genet. 2020 Mar;63(3):103768. doi: 10.1016/j.ejmg.2019.103768. Epub 2019 Sep 16. Eur J Med Genet. 2020. PMID: 31536828 Review.
-
Behavioral features in young adults with FG syndrome (Opitz-Kaveggia syndrome).Am J Med Genet C Semin Med Genet. 2010 Nov 15;154C(4):477-85. doi: 10.1002/ajmg.c.30284. Am J Med Genet C Semin Med Genet. 2010. PMID: 20981778 Free PMC article. Review.
Cited by
-
Clinical heterogeneity of Kabuki syndrome in a cohort of Italian patients and review of the literature.Eur J Pediatr. 2022 Jan;181(1):171-187. doi: 10.1007/s00431-021-04108-w. Epub 2021 Jul 7. Eur J Pediatr. 2022. PMID: 34232366 Free PMC article. Review.
-
Prenatal diagnosis in a fetuses with a clenched hands, overlapping fingers, and clubfoot due to MED12 deficiency in three affected siblings: A case report.Front Genet. 2023 Jul 12;14:1037345. doi: 10.3389/fgene.2023.1037345. eCollection 2023. Front Genet. 2023. PMID: 37501721 Free PMC article.
-
MED12 related disorders.Am J Med Genet A. 2013 Nov;161A(11):2734-40. doi: 10.1002/ajmg.a.36183. Epub 2013 Oct 10. Am J Med Genet A. 2013. PMID: 24123922 Free PMC article. Review.
-
Mediator and human disease.Semin Cell Dev Biol. 2011 Sep;22(7):776-87. doi: 10.1016/j.semcdb.2011.07.024. Epub 2011 Aug 4. Semin Cell Dev Biol. 2011. PMID: 21840410 Free PMC article. Review.
-
Potential roles of mediator Complex Subunit 13 in Cardiac Diseases.Int J Biol Sci. 2021 Jan 1;17(1):328-338. doi: 10.7150/ijbs.52290. eCollection 2021. Int J Biol Sci. 2021. PMID: 33390853 Free PMC article. Review.
References
-
- Opitz JM, Kaveggia EG. Studies of malformation syndromes of man 33: the FG syndrome. An X-linked recessive syndrome of multiple congenital anomalies and mental retardation. Z Kinderheilkd. 1974;117:1–18. - PubMed
-
- Risheg H, Graham JM, Jr, Clark RD, et al. A recurrent mutation in MED12 leading to R961W causes Opitz-Kaveggia syndrome. Nat Genet. 2007;39:451–453. - PubMed
-
- Lyons MJ, Graham JM, Jr, Neri G, et al. Clinical experience in the evaluation of 30 patients with a prior diagnosis of FG syndrome. J Med Genet. 2009;46:9–13. - PubMed
-
- Keller MA, Jones KL, Nyhan WL, Francke U, Dixson B. A new syndrome of mental deficiency with craniofacial, limb and anal abnormalities. J Pediatr. 1976;88:589 –591. - PubMed
-
- Riccardi VM, Hassler E, Lubinsky MS. The FG syndrome: further characterization, report of a third family, and of a sporadic case. Am J Med Genet. 1977;1:47–58. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
