

A novel matrix metalloproteinase 2 (MMP2) terminal hemopexin domain mutation in a family with multicentric osteolysis with nodulosis and arthritis with cardiac defects
- PMID: 18985071
- PMCID: PMC2721823
- DOI: 10.1038/ejhg.2008.204
A novel matrix metalloproteinase 2 (MMP2) terminal hemopexin domain mutation in a family with multicentric osteolysis with nodulosis and arthritis with cardiac defects
Abstract
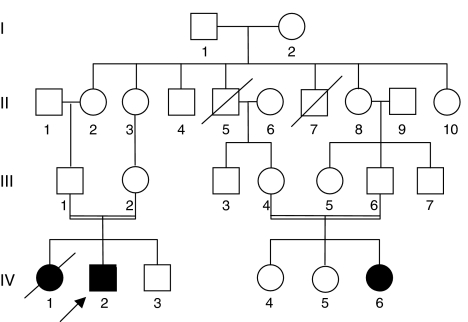
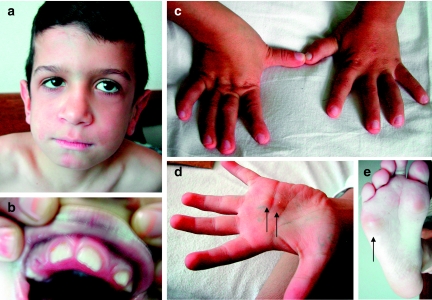
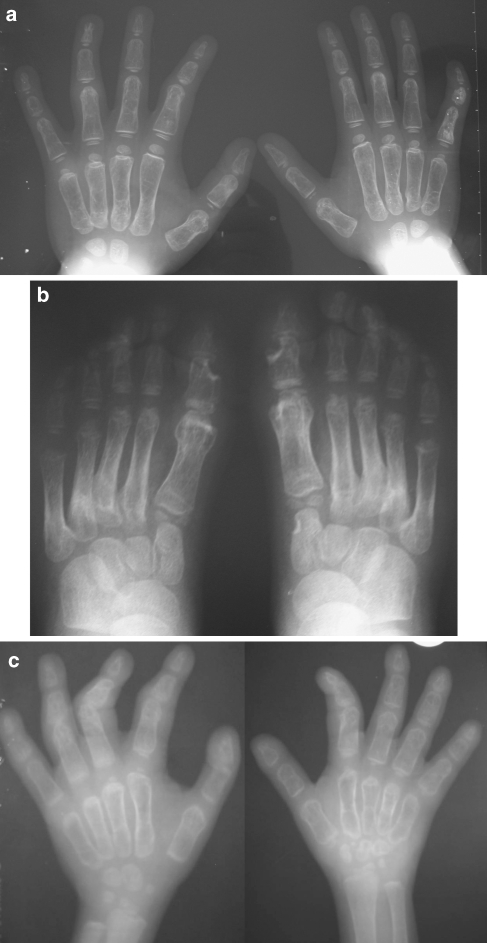
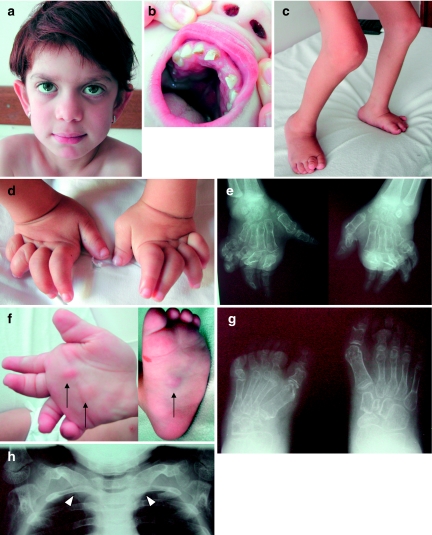
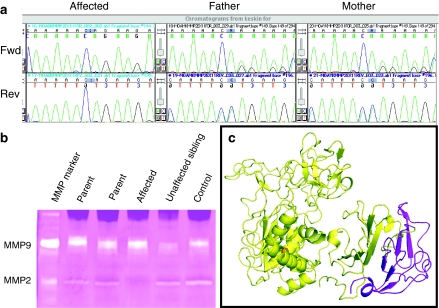
Multicentric osteolysis with nodulosis and arthropathy (MONA, NAO (OMIM no. 605156)) is an autosomal recessive member of the 'vanishing bone' syndromes and is notable for the extent of carpal and tarsal osteolysis and interphalangeal joint erosions, facial dysmorphia, and the presence of fibrocollagenous nodules. This rare disorder has been described previously in Saudi Arabian and Indian families. We now report on the first Turkish family with MONA, further confirming the panethnic nature of this disease. Strikingly, and in addition to the previously noted skeletal and joint features, affected members of this family also had congenital heart defects. Molecular analysis identified a novel MMP2 inactivating mutation that deletes the terminal hemopexin domains and thus confirmed the diagnosis of MONA. On the basis of these findings, we suggest that cardiac defects may also represent a component of this syndrome and thus a physiologically relevant target of MMP-2 activity.
Figures
Similar articles
-
Multicentric osteolysis with nodulosis and arthropathy (MONA) with cardiac malformation, mimicking polyarticular juvenile idiopathic arthritis: case report and literature review.Eur J Pediatr. 2013 Dec;172(12):1657-63. doi: 10.1007/s00431-013-2102-8. Epub 2013 Jul 31. Eur J Pediatr. 2013. PMID: 23900523 Review.
-
Mutation of the matrix metalloproteinase 2 gene (MMP2) causes a multicentric osteolysis and arthritis syndrome.Nat Genet. 2001 Jul;28(3):261-5. doi: 10.1038/90100. Nat Genet. 2001. PMID: 11431697
-
A novel mutation in the matrix metallopeptidase 2 coding gene associated with intrafamilial variability of multicentric osteolysis, nodulosis, and arthropathy.Mol Genet Genomic Med. 2019 Aug;7(8):e802. doi: 10.1002/mgg3.802. Epub 2019 Jul 3. Mol Genet Genomic Med. 2019. PMID: 31268248 Free PMC article.
-
Al-Aqeel Sewairi syndrome, a new autosomal recessive disorder with multicentric osteolysis, nodulosis and arthropathy. The first genetic defect of matrix metalloproteinase 2 gene.Saudi Med J. 2005 Jan;26(1):24-30. Saudi Med J. 2005. PMID: 15756348
-
Multicentric osteolytic syndromes represent a phenotypic spectrum defined by defective collagen remodeling.Am J Med Genet A. 2019 Aug;179(8):1652-1664. doi: 10.1002/ajmg.a.61264. Epub 2019 Jun 19. Am J Med Genet A. 2019. PMID: 31218820 Review.
Cited by
-
Patient with mutation in the matrix metalloproteinase 2 (MMP2) gene - a case report and review of the literature.J Clin Res Pediatr Endocrinol. 2014;6(1):40-6. doi: 10.4274/Jcrpe.1166. J Clin Res Pediatr Endocrinol. 2014. PMID: 24637309 Free PMC article. Review.
-
A novel gene mutation for multicentric osteolysis nodulosis and arthropathy: Case report and review of literature.Heliyon. 2023 Mar 24;9(4):e14865. doi: 10.1016/j.heliyon.2023.e14865. eCollection 2023 Apr. Heliyon. 2023. PMID: 37025869 Free PMC article.
-
Bisphosphonates in multicentric osteolysis, nodulosis and arthropathy (MONA) spectrum disorder - an alternative therapeutic approach.Sci Rep. 2016 Sep 30;6:34017. doi: 10.1038/srep34017. Sci Rep. 2016. PMID: 27687687 Free PMC article.
-
Clinical, radiographic and molecular characterization of two unrelated families with multicentric osteolysis, nodulosis, and arthropathy.BMC Musculoskelet Disord. 2023 Sep 14;24(1):735. doi: 10.1186/s12891-023-06856-2. BMC Musculoskelet Disord. 2023. PMID: 37710205 Free PMC article.
-
Destroy to Rebuild: The Connection Between Bone Tissue Remodeling and Matrix Metalloproteinases.Front Physiol. 2020 Feb 5;11:47. doi: 10.3389/fphys.2020.00047. eCollection 2020. Front Physiol. 2020. PMID: 32116759 Free PMC article. Review.
References
-
- Hardegger F, Simpson LA, Segmueller G. The syndrome of idiopathic osteolysis. classification, review, and case report. J Bone Joint Surg Br. 1985;67:88–93. - PubMed
-
- Petit P, Fryns JP. Distal osteolysis, short stature, mental retardation, and characteristic facial appearance: delineation of an autosomal recessive subtype of essential osteolysis. Am J Med Genet. 1986;25:537–541. - PubMed
-
- Al Aqeel A, Al Sewairi W, Edress B, Gorlin RJ, Desnick RJ, Martignetti JA. Inherited multicentric osteolysis with arthritis: a variant resembling torg syndrome in a saudi family. Am J Med Genet. 2000;93:11–18. - PubMed
-
- Al-Mayouf SM, Majeed M, Hugosson C, Bahabri S. New form of idiopathic osteolysis: nodulosis, arthropathy and osteolysis (NAO) syndrome. Am J Med Genet. 2000;93:5–10. - PubMed
-
- Hall CM. International nosology and classification of constitutional disorders of bone (2001) Am J Med Genet. 2002;113:65–77. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
