

ADCK3, an ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme Q10 deficiency
- PMID: 18319074
- PMCID: PMC2427193
- DOI: 10.1016/j.ajhg.2007.12.024
ADCK3, an ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme Q10 deficiency
Abstract
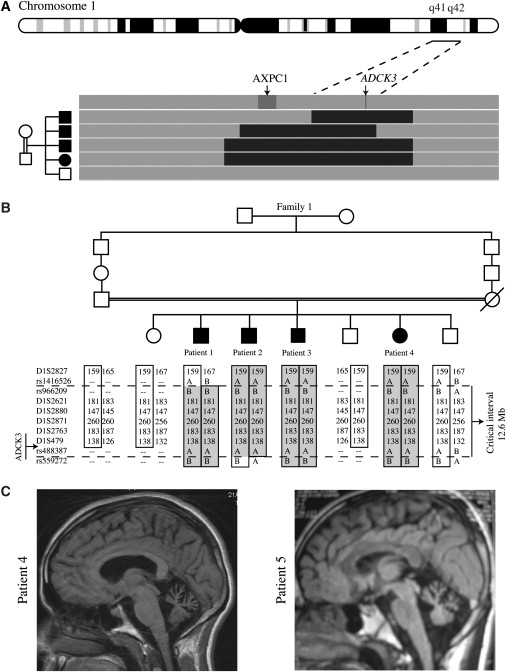
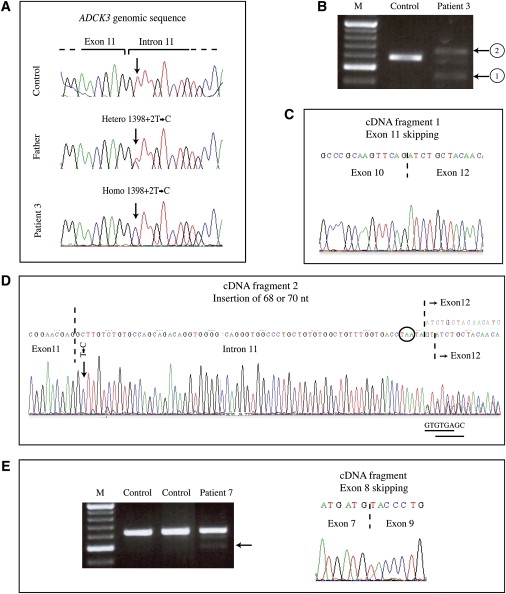
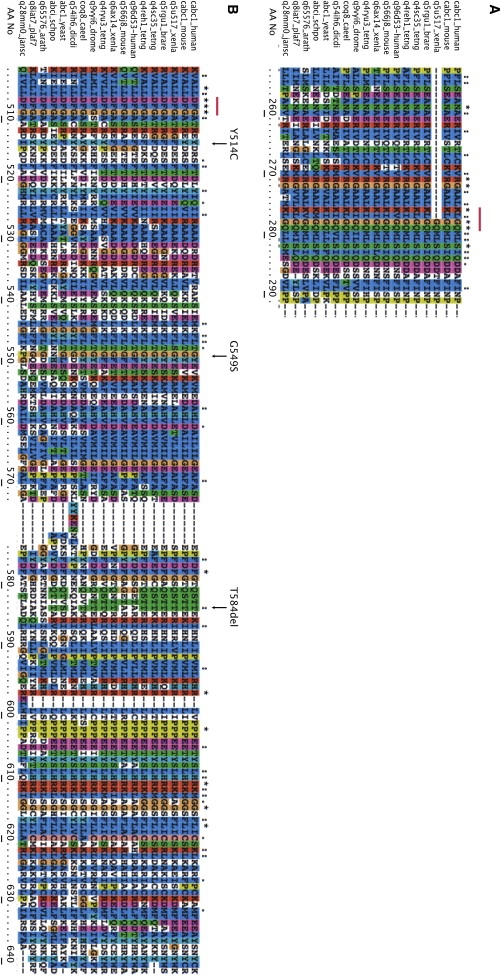
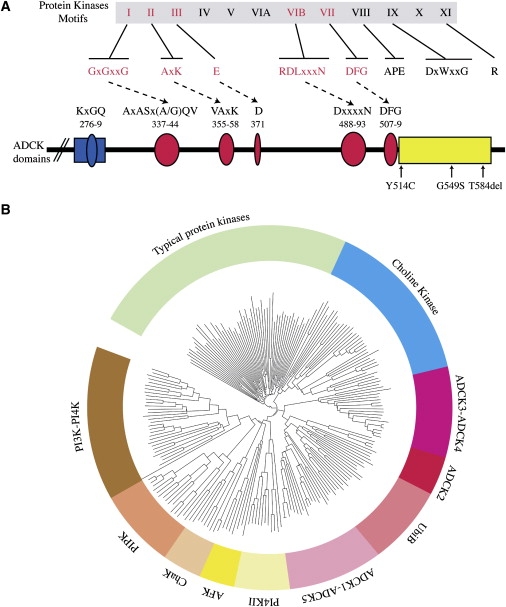
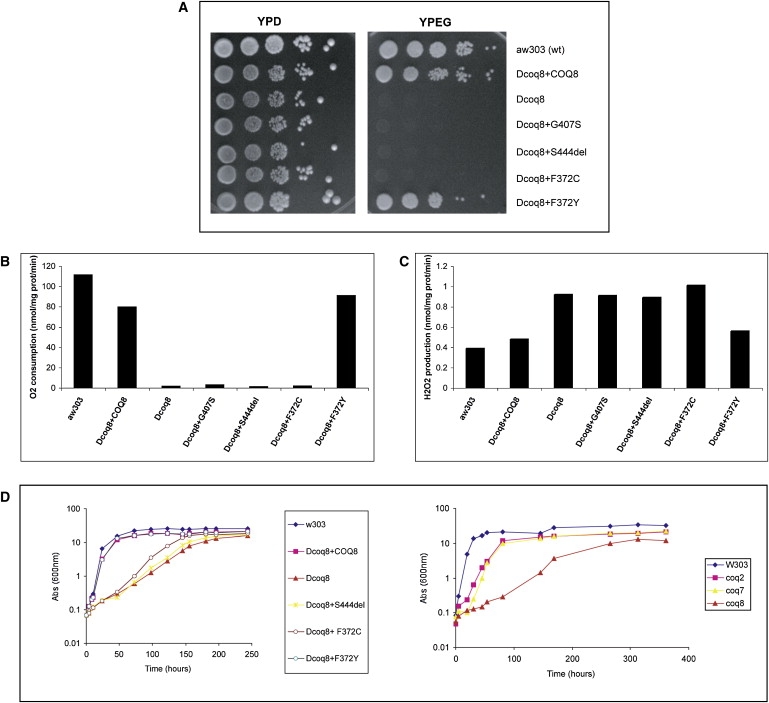
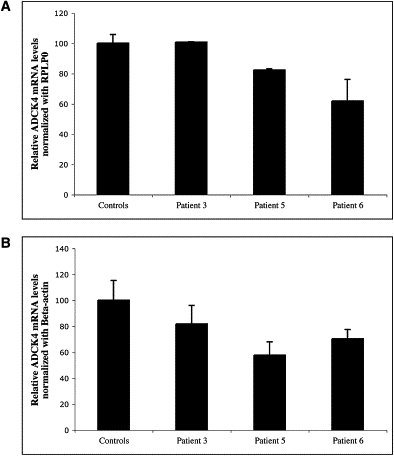
Muscle coenzyme Q(10) (CoQ(10) or ubiquinone) deficiency has been identified in more than 20 patients with presumed autosomal-recessive ataxia. However, mutations in genes required for CoQ(10) biosynthetic pathway have been identified only in patients with infantile-onset multisystemic diseases or isolated nephropathy. Our SNP-based genome-wide scan in a large consanguineous family revealed a locus for autosomal-recessive ataxia at chromosome 1q41. The causative mutation is a homozygous splice-site mutation in the aarF-domain-containing kinase 3 gene (ADCK3). Five additional mutations in ADCK3 were found in three patients with sporadic ataxia, including one known to have CoQ(10) deficiency in muscle. All of the patients have childhood-onset cerebellar ataxia with slow progression, and three of six have mildly elevated lactate levels. ADCK3 is a mitochondrial protein homologous to the yeast COQ8 and the bacterial UbiB proteins, which are required for CoQ biosynthesis. Three out of four patients tested showed a low endogenous pool of CoQ(10) in their fibroblasts or lymphoblasts, and two out of three patients showed impaired ubiquinone synthesis, strongly suggesting that ADCK3 is also involved in CoQ(10) biosynthesis. The deleterious nature of the three identified missense changes was confirmed by the introduction of them at the corresponding positions of the yeast COQ8 gene. Finally, a phylogenetic analysis shows that ADCK3 belongs to the family of atypical kinases, which includes phosphoinositide and choline kinases, suggesting that ADCK3 plays an indirect regulatory role in ubiquinone biosynthesis possibly as part of a feedback loop that regulates ATP production.
Figures
Similar articles
-
Primary Coenzyme Q deficiency Due to Novel ADCK3 Variants, Studies in Fibroblasts and Review of Literature.Neurochem Res. 2019 Oct;44(10):2372-2384. doi: 10.1007/s11064-019-02786-5. Epub 2019 Apr 9. Neurochem Res. 2019. PMID: 30968303
-
CABC1 gene mutations cause ubiquinone deficiency with cerebellar ataxia and seizures.Am J Hum Genet. 2008 Mar;82(3):623-30. doi: 10.1016/j.ajhg.2007.12.022. Am J Hum Genet. 2008. PMID: 18319072 Free PMC article.
-
Autosomal-recessive cerebellar ataxia caused by a novel ADCK3 mutation that elongates the protein: clinical, genetic and biochemical characterisation.J Neurol Neurosurg Psychiatry. 2014 May;85(5):493-8. doi: 10.1136/jnnp-2013-306483. Epub 2013 Nov 11. J Neurol Neurosurg Psychiatry. 2014. PMID: 24218524 Free PMC article.
-
Coenzyme Q biosynthesis in health and disease.Biochim Biophys Acta. 2016 Aug;1857(8):1079-1085. doi: 10.1016/j.bbabio.2016.03.036. Epub 2016 Apr 7. Biochim Biophys Acta. 2016. PMID: 27060254 Review.
-
Clinical, biochemical and molecular aspects of cerebellar ataxia and Coenzyme Q10 deficiency.Cerebellum. 2007;6(2):118-22. doi: 10.1080/14734220601021700. Cerebellum. 2007. PMID: 17510911 Review.
Cited by
-
Endophilin A2 Deficiency Impairs Antibody Production in Humans.J Clin Immunol. 2024 Nov 5;45(1):37. doi: 10.1007/s10875-024-01827-1. J Clin Immunol. 2024. PMID: 39499372
-
The podocyte power-plant disaster and its contribution to glomerulopathy.Front Endocrinol (Lausanne). 2014 Dec 15;5:209. doi: 10.3389/fendo.2014.00209. eCollection 2014. Front Endocrinol (Lausanne). 2014. PMID: 25566185 Free PMC article. Review.
-
COQ8A-Ataxia as a Manifestation of Primary Coenzyme Q Deficiency.Metabolites. 2022 Oct 8;12(10):955. doi: 10.3390/metabo12100955. Metabolites. 2022. PMID: 36295857 Free PMC article. Review.
-
Coenzyme Q protects Caenorhabditis elegans GABA neurons from calcium-dependent degeneration.Proc Natl Acad Sci U S A. 2010 Aug 10;107(32):14460-5. doi: 10.1073/pnas.0910630107. Epub 2010 Jul 27. Proc Natl Acad Sci U S A. 2010. PMID: 20663955 Free PMC article.
-
Neurodevelopmental manifestations of mitochondrial disease.J Dev Behav Pediatr. 2010 Sep;31(7):610-21. doi: 10.1097/DBP.0b013e3181ef42c1. J Dev Behav Pediatr. 2010. PMID: 20814259 Free PMC article. Review.
References
-
- Campuzano V., Montermini L., Molto M.D., Pianese L., Cossee M., Cavalcanti F., Monros E., Rodius F., Duclos F., Monticelli A. Friedreich's ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. - PubMed
-
- Allikmets R., Raskind W.H., Hutchinson A., Schueck N.D., Dean M., Koeller D.M. Mutation of a putative mitochondrial iron transporter gene (ABC7) in X-linked sideroblastic anemia and ataxia (XLSA/A) Hum. Mol. Genet. 1999;8:743–749. - PubMed
-
- Nikali K., Suomalainen A., Saharinen J., Kuokkanen M., Spelbrink J.N., Lonnqvist T., Peltonen L. Infantile onset spinocerebellar ataxia is caused by recessive mutations in mitochondrial proteins Twinkle and Twinky. Hum. Mol. Genet. 2005;14:2981–2990. - PubMed
-
- Van Goethem G., Martin J.J., Dermaut B., Lofgren A., Wibail A., Ververken D., Tack P., Dehaene I., Van Zandijcke M., Moonen M. Recessive POLG mutations presenting with sensory and ataxic neuropathy in compound heterozygote patients with progressive external ophthalmoplegia. Neuromuscul. Disord. 2003;13:133–142. - PubMed
-
- Winterthun S., Ferrari G., He L., Taylor R.W., Zeviani M., Turnbull D.M., Engelsen B.A., Moen G., Bindoff L.A. Autosomal recessive mitochondrial ataxic syndrome due to mitochondrial polymerase gamma mutations. Neurology. 2005;64:1204–1208. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
