

CABC1 gene mutations cause ubiquinone deficiency with cerebellar ataxia and seizures
- PMID: 18319072
- PMCID: PMC2427298
- DOI: 10.1016/j.ajhg.2007.12.022
CABC1 gene mutations cause ubiquinone deficiency with cerebellar ataxia and seizures
Abstract
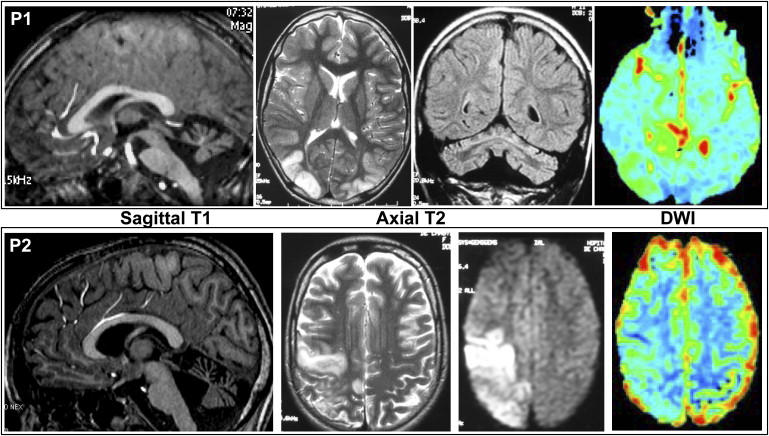
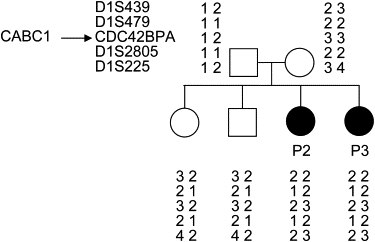
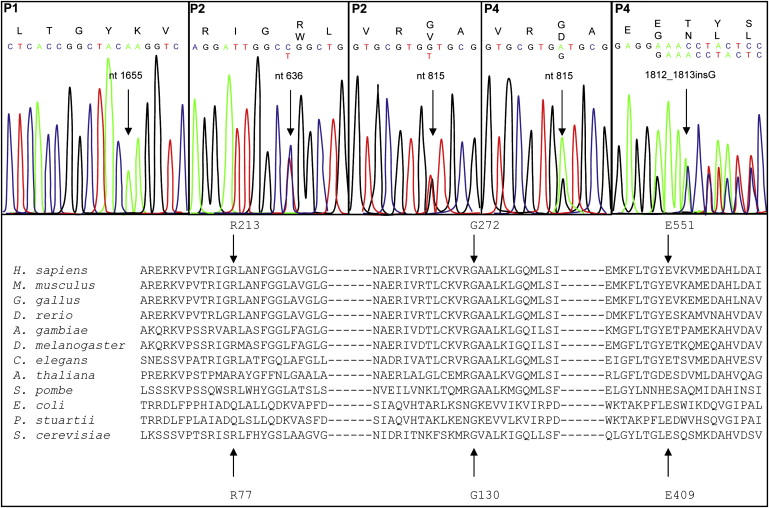
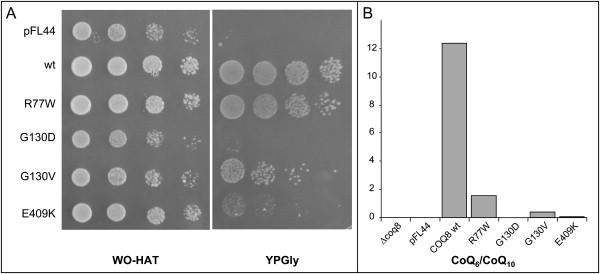
Coenzyme Q(10) (CoQ(10)) plays a pivotal role in oxidative phosphorylation (OXPHOS) in that it distributes electrons between the various dehydrogenases and the cytochrome segments of the respiratory chain. Primary coenzyme Q(10) deficiency represents a clinically heterogeneous condition suggestive of genetic heterogeneity, and several disease genes have been previously identified. The CABC1 gene, also called COQ8 or ADCK3, is the human homolog of the yeast ABC1/COQ8 gene, one of the numerous genes involved in the ubiquinone biosynthesis pathway. The exact function of the Abc1/Coq8 protein is as yet unknown, but this protein is classified as a putative protein kinase. We report here CABC1 gene mutations in four ubiquinone-deficient patients in three distinct families. These patients presented a similar progressive neurological disorder with cerebellar atrophy and seizures. In all cases, enzymological studies pointed to ubiquinone deficiency. CoQ(10) deficiency was confirmed by decreased content of ubiquinone in muscle. Various missense mutations (R213W, G272V, G272D, and E551K) modifying highly conserved amino acids of the protein and a 1 bp frameshift insertion c.[1812_1813insG] were identified. The missense mutations were introduced into the yeast ABC1/COQ8 gene and expressed in a Saccharomyces cerevisiae strain in which the ABC1/COQ8 gene was deleted. All the missense mutations resulted in a respiratory phenotype with no or decreased growth on glycerol medium and a severe reduction in ubiquinone synthesis, demonstrating that these mutations alter the protein function.
Figures
Similar articles
-
ADCK3, an ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme Q10 deficiency.Am J Hum Genet. 2008 Mar;82(3):661-72. doi: 10.1016/j.ajhg.2007.12.024. Am J Hum Genet. 2008. PMID: 18319074 Free PMC article.
-
Expression of the human atypical kinase ADCK3 rescues coenzyme Q biosynthesis and phosphorylation of Coq polypeptides in yeast coq8 mutants.Biochim Biophys Acta. 2011 May;1811(5):348-60. doi: 10.1016/j.bbalip.2011.01.009. Epub 2011 Feb 4. Biochim Biophys Acta. 2011. PMID: 21296186 Free PMC article.
-
A defect in coenzyme Q biosynthesis is responsible for the respiratory deficiency in Saccharomyces cerevisiae abc1 mutants.J Biol Chem. 2001 May 25;276(21):18161-8. doi: 10.1074/jbc.M100952200. Epub 2001 Mar 9. J Biol Chem. 2001. PMID: 11279158
-
Coenzyme Q biosynthesis in health and disease.Biochim Biophys Acta. 2016 Aug;1857(8):1079-1085. doi: 10.1016/j.bbabio.2016.03.036. Epub 2016 Apr 7. Biochim Biophys Acta. 2016. PMID: 27060254 Review.
-
Clinical, biochemical and molecular aspects of cerebellar ataxia and Coenzyme Q10 deficiency.Cerebellum. 2007;6(2):118-22. doi: 10.1080/14734220601021700. Cerebellum. 2007. PMID: 17510911 Review.
Cited by
-
Update on clinical aspects and treatment of selected vitamin-responsive disorders II (riboflavin and CoQ 10).J Inherit Metab Dis. 2012 Jul;35(4):679-87. doi: 10.1007/s10545-011-9434-1. Epub 2012 Jan 10. J Inherit Metab Dis. 2012. PMID: 22231380 Review.
-
Arabidopsis atypical kinase ABC1K1 is involved in red light-mediated development.Plant Cell Rep. 2016 Jun;35(6):1213-20. doi: 10.1007/s00299-016-1953-7. Epub 2016 Apr 2. Plant Cell Rep. 2016. PMID: 27038938
-
Mitochondrial function and lifespan of mice with controlled ubiquinone biosynthesis.Nat Commun. 2015 Mar 6;6:6393. doi: 10.1038/ncomms7393. Nat Commun. 2015. PMID: 25744659 Free PMC article.
-
Ataxia due to a COQ8A Novel Variant in Primary Coenzyme Q10 Deficiency.Mov Disord Clin Pract. 2023 Aug 24;10(Suppl 3):S41-S44. doi: 10.1002/mdc3.13781. eCollection 2023 Aug. Mov Disord Clin Pract. 2023. PMID: 37636224 Free PMC article. No abstract available.
-
Animal Models of Coenzyme Q Deficiency: Mechanistic and Translational Learnings.Antioxidants (Basel). 2021 Oct 26;10(11):1687. doi: 10.3390/antiox10111687. Antioxidants (Basel). 2021. PMID: 34829558 Free PMC article. Review.
References
-
- Mollet J., Giurgea I., Schlemmer D., Dallner G., Chretien D., Delahodde A., Bacq D., de Lonlay P., Munnich A., Rötig A. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J. Clin. Invest. 2007;117:765–772. - PMC - PubMed
-
- Diomedi-Camassei F., Di Giandomenico S., Santorelli F.M., Caridi G., Piemonte F., Montini G., Ghiggeri G.M., Murer L., Barisoni L., Pastore A. COQ2 nephropathy: A newly described inherited mitochondriopathy with primary renal involvement. J. Am. Soc. Nephrol. 2007;18:2773–2780. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
