

Characterization of Potocki-Lupski syndrome (dup(17)(p11.2p11.2)) and delineation of a dosage-sensitive critical interval that can convey an autism phenotype
- PMID: 17357070
- PMCID: PMC1852712
- DOI: 10.1086/512864
Characterization of Potocki-Lupski syndrome (dup(17)(p11.2p11.2)) and delineation of a dosage-sensitive critical interval that can convey an autism phenotype
Abstract
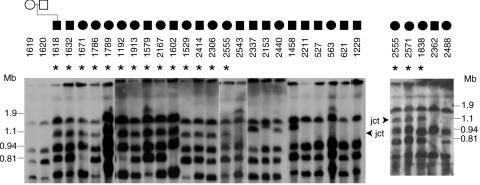
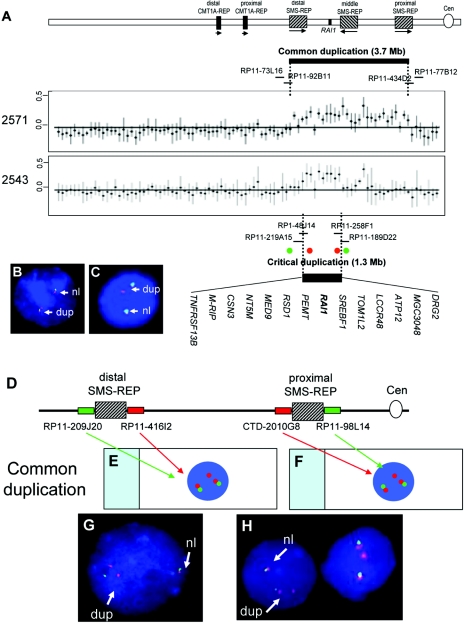
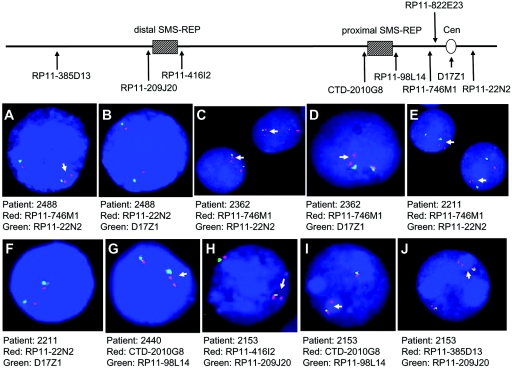
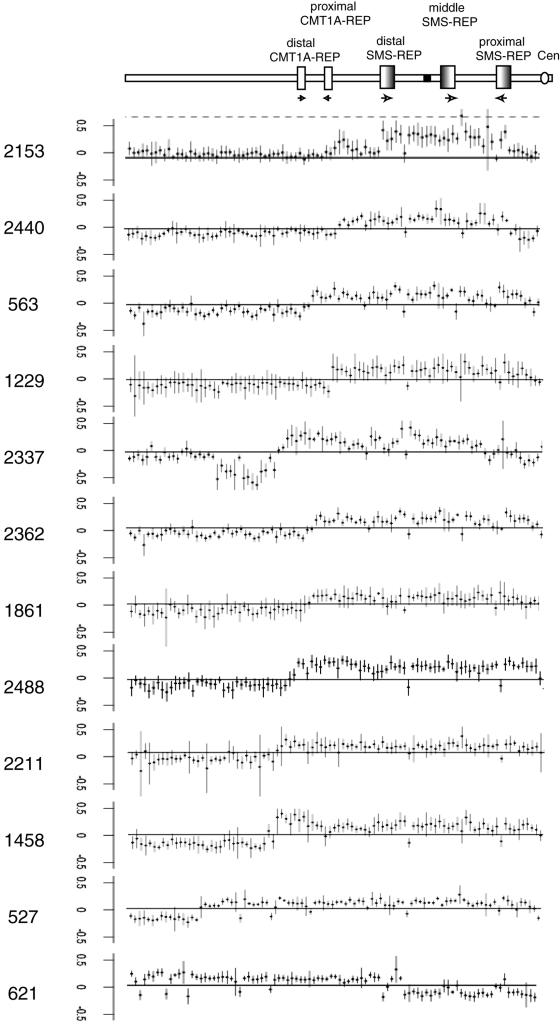
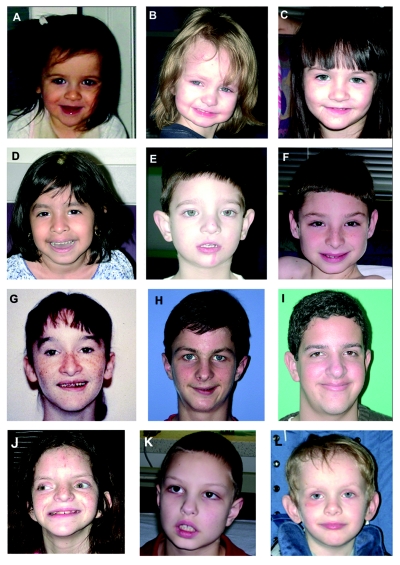
The duplication 17p11.2 syndrome, associated with dup(17)(p11.2p11.2), is a recently recognized syndrome of multiple congenital anomalies and mental retardation and is the first predicted reciprocal microduplication syndrome described--the homologous recombination reciprocal of the Smith-Magenis syndrome (SMS) microdeletion (del(17)(p11.2p11.2)). We previously described seven subjects with dup(17)(p11.2p11.2) and noted their relatively mild phenotype compared with that of individuals with SMS. Here, we molecularly analyzed 28 additional patients, using multiple independent assays, and also report the phenotypic characteristics obtained from extensive multidisciplinary clinical study of a subset of these patients. Whereas the majority of subjects (22 of 35) harbor the homologous recombination reciprocal product of the common SMS microdeletion (~3.7 Mb), 13 subjects (~37%) have nonrecurrent duplications ranging in size from 1.3 to 15.2 Mb. Molecular studies suggest potential mechanistic differences between nonrecurrent duplications and nonrecurrent genomic deletions. Clinical features observed in patients with the common dup(17)(p11.2p11.2) are distinct from those seen with SMS and include infantile hypotonia, failure to thrive, mental retardation, autistic features, sleep apnea, and structural cardiovascular anomalies. We narrow the critical region to a 1.3-Mb genomic interval that contains the dosage-sensitive RAI1 gene. Our results refine the critical region for Potocki-Lupski syndrome, provide information to assist in clinical diagnosis and management, and lend further support for the concept that genomic architecture incites genomic instability.
Figures


Similar articles
-
Inherited dup(17)(p11.2p11.2): expanding the phenotype of the Potocki-Lupski syndrome.Am J Med Genet A. 2014 Feb;164A(2):500-4. doi: 10.1002/ajmg.a.36287. Epub 2013 Dec 5. Am J Med Genet A. 2014. PMID: 24311450 Review.
-
Molecular mechanism for duplication 17p11.2- the homologous recombination reciprocal of the Smith-Magenis microdeletion.Nat Genet. 2000 Jan;24(1):84-7. doi: 10.1038/71743. Nat Genet. 2000. PMID: 10615134
-
Potocki-Lupski syndrome: an inherited dup(17)(p11.2p11.2) with hypoplastic left heart.Am J Med Genet A. 2011 Feb;155A(2):367-71. doi: 10.1002/ajmg.a.33845. Am J Med Genet A. 2011. PMID: 21271656
-
Variability in clinical phenotype despite common chromosomal deletion in Smith-Magenis syndrome [del(17)(p11.2p11.2)].Genet Med. 2003 Nov-Dec;5(6):430-4. doi: 10.1097/01.gim.0000095625.14160.ab. Genet Med. 2003. PMID: 14614393
-
Trisomy 17p10-p12 due to mosaic supernumerary marker chromosome: delineation of molecular breakpoints and clinical phenotype, and comparison to other proximal 17p segmental duplications.Am J Med Genet A. 2005 Oct 1;138A(2):175-80. doi: 10.1002/ajmg.a.30948. Am J Med Genet A. 2005. PMID: 16152635 Review.
Cited by
-
Objective measures of sleep disturbances in children with Potocki-Lupski syndrome.Am J Med Genet A. 2019 Oct;179(10):1982-1986. doi: 10.1002/ajmg.a.61307. Epub 2019 Jul 24. Am J Med Genet A. 2019. PMID: 31342617 Free PMC article.
-
Evaluation of genetic variants using chromosomal microarray analysis for fetuses with polyhydramnios.BMC Med Genomics. 2022 Mar 30;15(1):73. doi: 10.1186/s12920-022-01224-w. BMC Med Genomics. 2022. PMID: 35354480 Free PMC article.
-
Phenotypic variability and genetic susceptibility to genomic disorders.Hum Mol Genet. 2010 Oct 15;19(R2):R176-87. doi: 10.1093/hmg/ddq366. Epub 2010 Aug 31. Hum Mol Genet. 2010. PMID: 20807775 Free PMC article. Review.
-
Case Report: Potocki-Lupski Syndrome in Five Siblings.Front Pediatr. 2021 Nov 8;9:698629. doi: 10.3389/fped.2021.698629. eCollection 2021. Front Pediatr. 2021. PMID: 34820340 Free PMC article.
-
Genomewide association study for C-reactive protein in Indians replicates known associations of common variants.J Genet. 2019 Mar;98:20. J Genet. 2019. PMID: 30945673
References
Web Resources
-
- Database of Genomic Variants, http://projects.tcag.ca/variation/ (for Human Genome Assembly build 36)
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for CMT1A, HNPP, SMS, RAI1, PMP22, and PLP1)
-
- Segmental Duplication Database, http://humanparalogy.gs.washington.edu/
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
