

A novel COL1A1 mutation in infantile cortical hyperostosis (Caffey disease) expands the spectrum of collagen-related disorders
- PMID: 15864348
- PMCID: PMC1087158
- DOI: 10.1172/JCI22760
A novel COL1A1 mutation in infantile cortical hyperostosis (Caffey disease) expands the spectrum of collagen-related disorders
Abstract
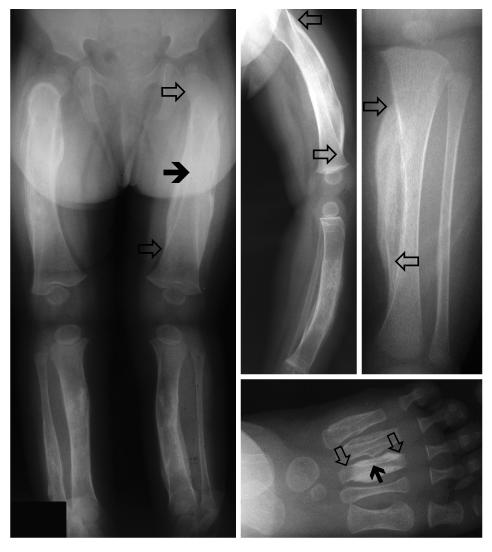
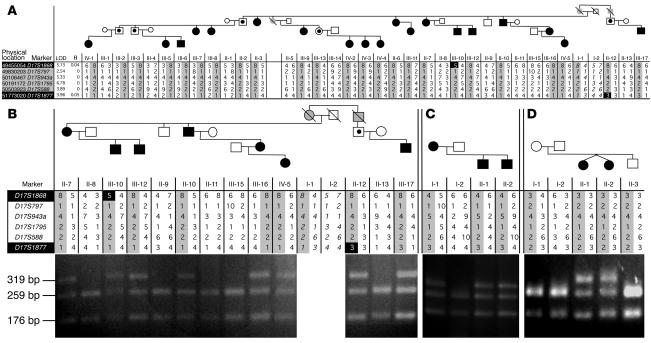
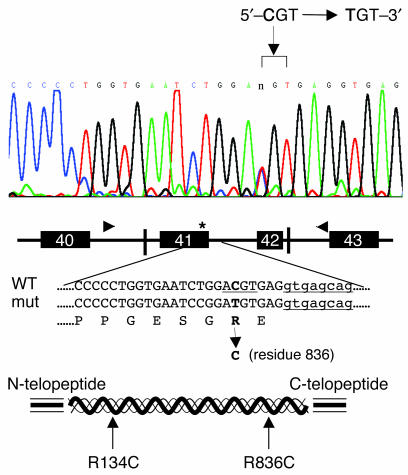
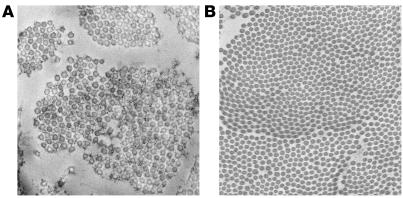
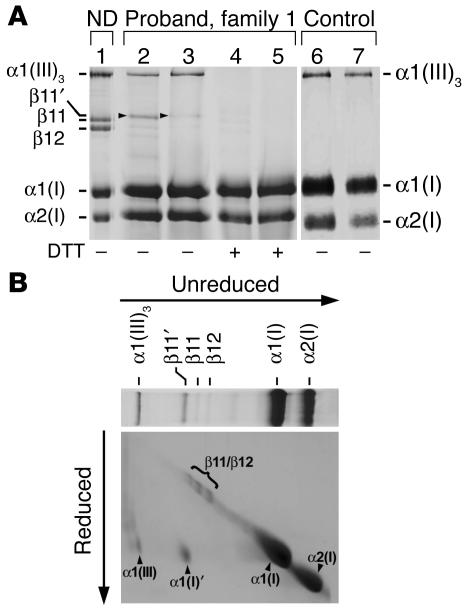
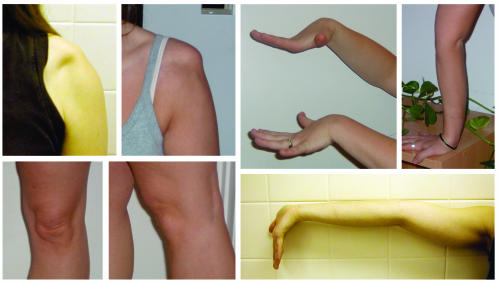
Infantile cortical hyperostosis (Caffey disease) is characterized by spontaneous episodes of subperiosteal new bone formation along 1 or more bones commencing within the first 5 months of life. A genome-wide screen for genetic linkage in a large family with an autosomal dominant form of Caffey disease (ADC) revealed a locus on chromosome 17q21 (LOD score, 6.78). Affected individuals and obligate carriers were heterozygous for a missense mutation (3040Ctwo head right arrowT) in exon 41 of the gene encoding the alpha1(I) chain of type I collagen (COL1A1), altering residue 836 (R836C) in the triple-helical domain of this chain. The same mutation was identified in affected members of 2 unrelated, smaller families with ADC, but not in 2 prenatal cases and not in more than 300 chromosomes from healthy individuals. Fibroblast cultures from an affected individual produced abnormal disulfide-bonded dimeric alpha1(I) chains. Dermal collagen fibrils of the same individual were larger, more variable in shape and size, and less densely packed than those in control samples. Individuals bearing the mutation, whether they had experienced an episode of cortical hyperostosis or not, had joint hyperlaxity, hyperextensible skin, and inguinal hernias resembling symptoms of a mild form of Ehlers-Danlos syndrome type III. These findings extend the spectrum of COL1A1-related diseases to include a hyperostotic disorder.
Figures
Comment in
-
Caffey disease: an unlikely collagenopathy.J Clin Invest. 2005 May;115(5):1142-4. doi: 10.1172/JCI25148. J Clin Invest. 2005. PMID: 15864344 Free PMC article.
Similar articles
-
Caffey disease: an unlikely collagenopathy.J Clin Invest. 2005 May;115(5):1142-4. doi: 10.1172/JCI25148. J Clin Invest. 2005. PMID: 15864344 Free PMC article.
-
Expanding the phenotypic spectrum of Caffey disease.Clin Genet. 2007 Mar;71(3):280-4. doi: 10.1111/j.1399-0004.2007.00768.x. Clin Genet. 2007. PMID: 17309652
-
The c.3040C > T mutation in COL1A1 is recurrent in Korean patients with infantile cortical hyperostosis (Caffey disease).J Hum Genet. 2008;53(10):947. doi: 10.1007/s10038-008-0328-5. Epub 2008 Aug 13. J Hum Genet. 2008. PMID: 18704262
-
Two Japanese familial cases of Caffey disease with and without the common COL1A1 mutation and normal bone density, and review of the literature.Eur J Pediatr. 2014 Jun;173(6):799-804. doi: 10.1007/s00431-013-2252-8. Epub 2014 Jan 4. Eur J Pediatr. 2014. PMID: 24390061 Review.
-
Recurrence of infantile cortical hyperostosis: a case report and review of the literature.J Pediatr Orthop. 2013 Mar;33(2):e10-7. doi: 10.1097/BPO.0b013e318277d3a2. J Pediatr Orthop. 2013. PMID: 23389580 Review.
Cited by
-
Degenerative suspensory ligament desmitis as a systemic disorder characterized by proteoglycan accumulation.BMC Vet Res. 2006 Apr 12;2:12. doi: 10.1186/1746-6148-2-12. BMC Vet Res. 2006. PMID: 16611357 Free PMC article.
-
COL1A1 mutation in an Indian child with Caffey disease.Indian J Pediatr. 2011 Jul;78(7):877-9. doi: 10.1007/s12098-010-0339-z. Epub 2011 Jan 20. Indian J Pediatr. 2011. PMID: 21249479
-
The Genetic Architecture of High Bone Mass.Front Endocrinol (Lausanne). 2020 Oct 29;11:595653. doi: 10.3389/fendo.2020.595653. eCollection 2020. Front Endocrinol (Lausanne). 2020. PMID: 33193107 Free PMC article. Review.
-
Caffey's disease in disguise: a child abuse mimic.BMJ Case Rep. 2024 Feb 7;17(2):e256998. doi: 10.1136/bcr-2023-256998. BMJ Case Rep. 2024. PMID: 38331449
-
The triple helix of collagens - an ancient protein structure that enabled animal multicellularity and tissue evolution.J Cell Sci. 2018 Apr 9;131(7):jcs203950. doi: 10.1242/jcs.203950. J Cell Sci. 2018. PMID: 29632050 Free PMC article. Review.
References
-
- Bernstein RM, Zaleske DJ. Familial aspects of Caffey’s disease. Am. J. Orthop. 1995;24:777–781. - PubMed
-
- Caffey J. On some late skeletal changes in chronic infantile cortical hyperostosis. Radiology. 1952;59:651–657. - PubMed
-
- Blank E. Recurrent Caffey’s cortical hyperostosis and persistent deformity. Pediatrics. 1975;55:856–860. - PubMed
-
- Heyman E, Laver J, Beer S. Prostaglandin synthetase inhibitor in Caffey disease. J. Pediatr. 1982;101:314. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
