

Mutations in the gene encoding gap junction protein alpha 12 (connexin 46.6) cause Pelizaeus-Merzbacher-like disease
- PMID: 15192806
- PMCID: PMC1216059
- DOI: 10.1086/422763
Mutations in the gene encoding gap junction protein alpha 12 (connexin 46.6) cause Pelizaeus-Merzbacher-like disease
Erratum in
- Am J Hum Genet. 2004 Oct;5(4):737
Abstract
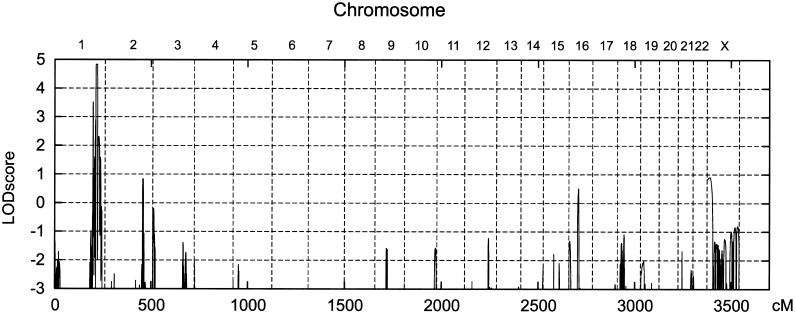
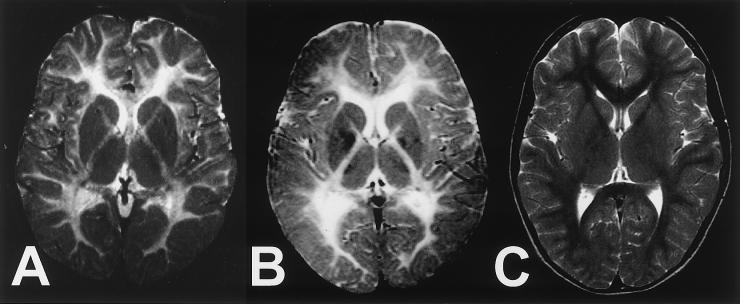
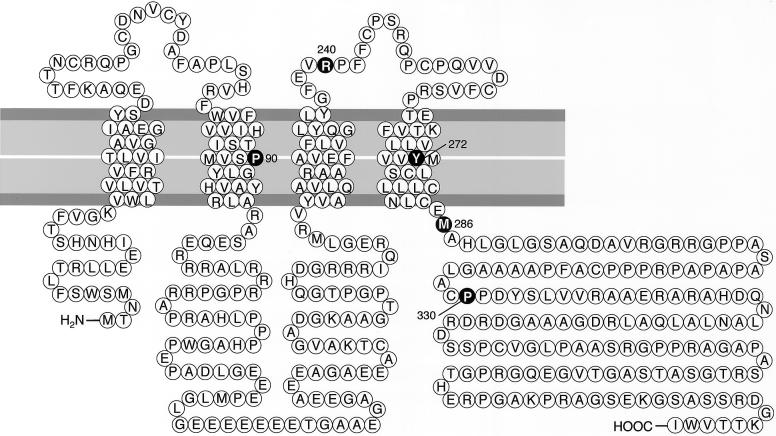
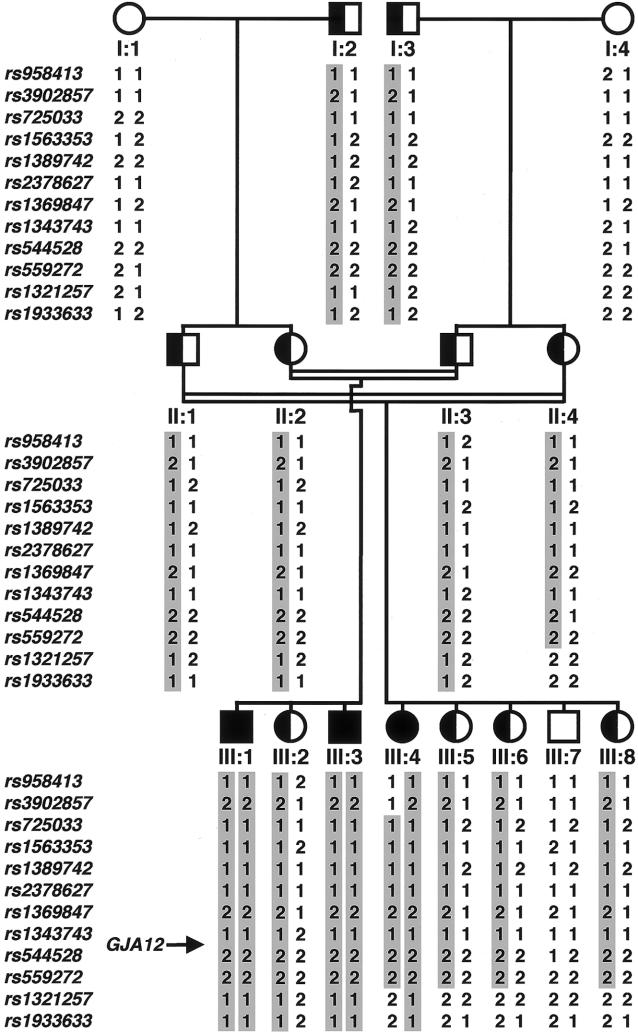
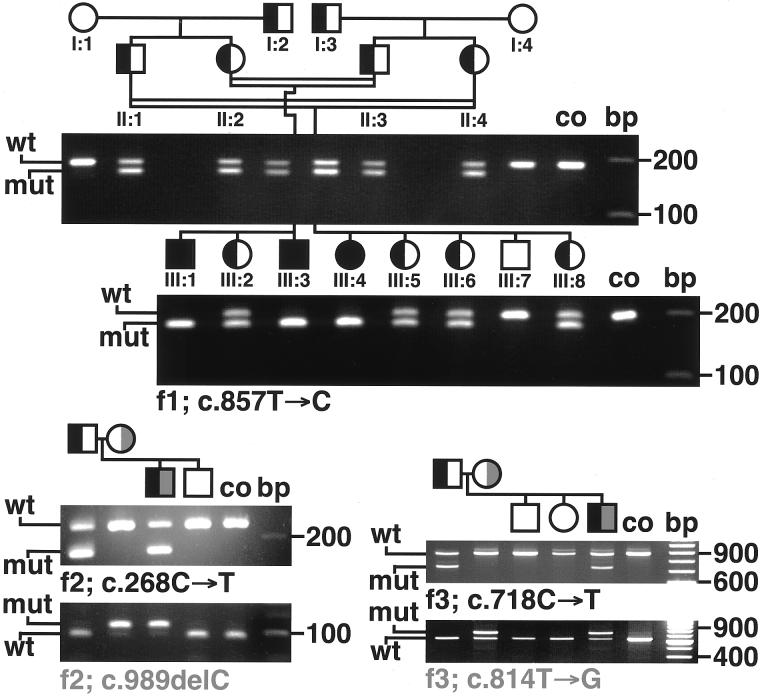
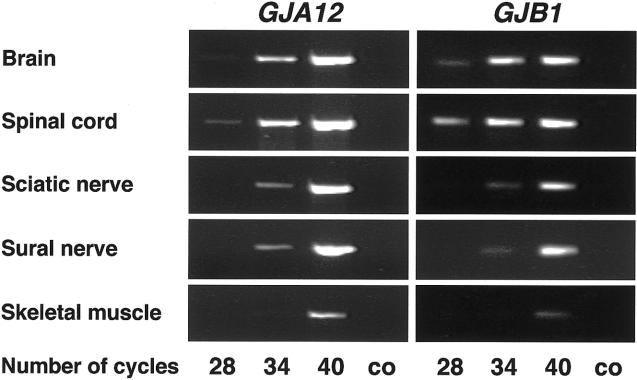
The hypomyelinating leukodystrophies X-linked Pelizaeus-Merzbacher disease (PMD) and Pelizaeus-Merzbacher-like disease (PMLD) are characterized by nystagmus, progressive spasticity, and ataxia. In a consanguineous family with PMLD, we performed a genomewide linkage scan using the GeneChip Mapping EA 10K Array (Affymetrix) and detected a single gene locus on chromosome 1q41-q42. This region harbors the GJA12 gene, which encodes gap junction protein alpha 12 (or connexin 46.6). Gap junction proteins assemble into intercellular channels through which signaling ions and small molecules are exchanged. GJA12 is highly expressed in oligodendrocytes, and, therefore, it serves as an excellent candidate for hypomyelination in PMLD. In three of six families with PMLD, we detected five different GJA12 mutations, including missense, nonsense, and frameshift mutations. We thereby confirm previous assumptions that PMLD is genetically heterogeneous. Although the murine Gja12 ortholog is not expressed in sciatic nerve, we did detect GJA12 transcripts in human sciatic and sural nerve tissue by reverse-transcriptase polymerase chain reaction. These results are in accordance with the electrophysiological finding of reduced motor and sensory nerve conduction velocities in patients with PMLD, which argues for a demyelinating neuropathy. In this study, we demonstrate that GJA12 plays a key role in central myelination and is involved in peripheral myelination in humans.
Figures

Similar articles
-
Clinical neurophysiology in GJA12-related hypomyelination vs Pelizaeus-Merzbacher disease.Neurology. 2010 Jun 1;74(22):1785-9. doi: 10.1212/WNL.0b013e3181e0f820. Neurology. 2010. PMID: 20513814
-
GJA12 mutations are a rare cause of Pelizaeus-Merzbacher-like disease.Neurology. 2008 Mar 4;70(10):748-54. doi: 10.1212/01.wnl.0000284828.84464.35. Epub 2007 Dec 19. Neurology. 2008. PMID: 18094336
-
GJA12 mutations in children with recessive hypomyelinating leukoencephalopathy.Neurology. 2006 Jul 25;67(2):273-9. doi: 10.1212/01.wnl.0000223832.66286.e4. Epub 2006 May 17. Neurology. 2006. PMID: 16707726 Clinical Trial.
-
Pelizaeus-Merzbacher disease, Pelizaeus-Merzbacher-like disease 1, and related hypomyelinating disorders.Semin Neurol. 2012 Feb;32(1):62-7. doi: 10.1055/s-0032-1306388. Epub 2012 Mar 15. Semin Neurol. 2012. PMID: 22422208 Review.
-
Pelizaeus-Merzbacher Disease: Molecular and Cellular Pathologies and Associated Phenotypes.Adv Exp Med Biol. 2019;1190:201-216. doi: 10.1007/978-981-32-9636-7_13. Adv Exp Med Biol. 2019. PMID: 31760646 Review.
Cited by
-
Association of the HLA region with multiple sclerosis as confirmed by a genome screen using >10,000 SNPs on DNA chips.J Mol Med (Berl). 2005 Jun;83(6):486-94. doi: 10.1007/s00109-005-0650-8. Epub 2005 Mar 16. J Mol Med (Berl). 2005. PMID: 15770496
-
Magnetic resonance imaging pattern recognition in hypomyelinating disorders.Brain. 2010 Oct;133(10):2971-82. doi: 10.1093/brain/awq257. Brain. 2010. PMID: 20881161 Free PMC article.
-
Connexins and Disease.Cold Spring Harb Perspect Biol. 2018 Sep 4;10(9):a029348. doi: 10.1101/cshperspect.a029348. Cold Spring Harb Perspect Biol. 2018. PMID: 28778872 Free PMC article. Review.
-
Gene therapy targeting oligodendrocytes provides therapeutic benefit in a leukodystrophy model.Brain. 2017 Mar 1;140(3):599-616. doi: 10.1093/brain/aww351. Brain. 2017. PMID: 28100454 Free PMC article.
-
Microtubule-assisted altered trafficking of astrocytic gap junction protein connexin 43 is associated with depletion of connexin 47 during mouse hepatitis virus infection.J Biol Chem. 2017 Sep 8;292(36):14747-14763. doi: 10.1074/jbc.M117.786491. Epub 2017 May 31. J Biol Chem. 2017. PMID: 28566289 Free PMC article.
References
Electronic-Database Information
-
- dbSNP Home Page, http://www.ncbi.nlm.nih.gov/SNP/ (for refSNP ID rs958413, rs3902857, rs725033, rs1563353, rs1389742, rs2378627, rs1369847, rs1343743, rs544528, rs559272, rs1321257, and rs1933633)
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for human GJA12 mRNA [accession number NM_020435], human GJA12 genomic sequence [accession number NT_004559], mouse Gja12 mRNA [accession number NM_080454], Gja12_fugu [accession number CAAB01002259.1], human GJB1 mRNA [accession number NM_000166], human GJB1 genomic sequence [accession number NT_011669])
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for CMTX1 [MIM #302800], PMD [MIM #312080], PMLD [MIM 311601])
-
- UniProt/TrEMBL, http://www.ebi.ac.uk/trembl/ (for Gja12_mouse [accession number Q9EPM1] and Gja12_rat [accession number Q80XF7])
References
-
- Bergoffen J, Scherer SS, Wang S, Scott MO, Bone LJ, Paul DL, Chen K, Lensch MW, Chance PF, Fischbeck KH (1993) Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science 262:2039–2042 - PubMed
-
- Garbern JY, Cambi F, Tang XM, Sima AA, Vallat JM, Bosch EP, Lewis R, Shy M, Sohi J, Kraft G, Chen KL, Joshi I, Leonard DG, Johnson W, Raskind W, Dlouhy SR, Pratt V, Hodes ME, Bird T, Kamholz J (1997) Proteolipid protein is necessary in peripheral as well as central myelin. Neuron 19:205–21810.1016/S0896-6273(00)80360-8 - DOI - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
