

Mutations of ephrin-B1 (EFNB1), a marker of tissue boundary formation, cause craniofrontonasal syndrome
- PMID: 15166289
- PMCID: PMC423250
- DOI: 10.1073/pnas.0402819101
Mutations of ephrin-B1 (EFNB1), a marker of tissue boundary formation, cause craniofrontonasal syndrome
Abstract
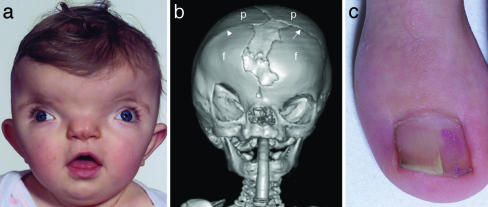
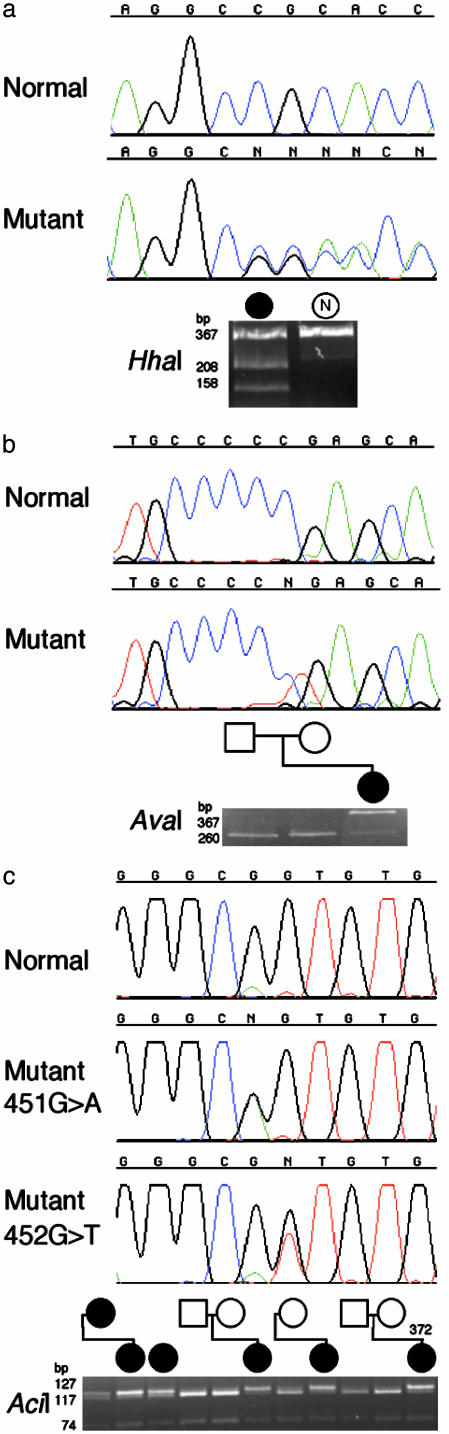
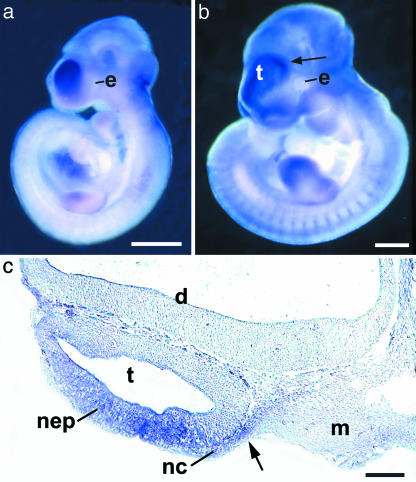
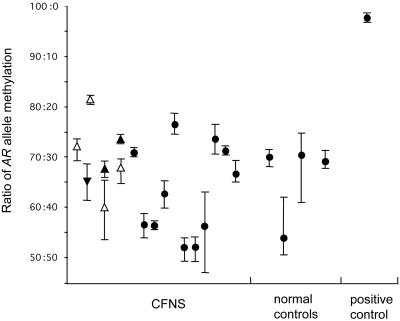
Craniofrontonasal syndrome (CFNS) is an X-linked developmental disorder that shows paradoxically greater severity in heterozygous females than in hemizygous males. Females have frontonasal dysplasia and coronal craniosynostosis (fusion of the coronal sutures); in males, hypertelorism is the only typical manifestation. Here, we show that the classical female CFNS phenotype is caused by heterozygous loss-of-function mutations in EFNB1, which encodes a member of the ephrin family of transmembrane ligands for Eph receptor tyrosine kinases. In mice, the orthologous Efnb1 gene is expressed in the frontonasal neural crest and demarcates the position of the future coronal suture. Although EFNB1 is X-inactivated, we did not observe markedly skewed X-inactivation in either blood or cranial periosteum from females with CFNS, indicating that lack of ephrin-B1 does not compromise cell viability in these tissues. We propose that in heterozygous females, patchwork loss of ephrin-B1 disturbs tissue boundary formation at the developing coronal suture, whereas in males deficient in ephrin-B1, an alternative mechanism maintains the normal boundary. This is the only known mutation in the ephrin/Eph receptor signaling system in humans and provides clues to the biogenesis of craniosynostosis.
Figures

Similar articles
-
Twenty-six novel EFNB1 mutations in familial and sporadic craniofrontonasal syndrome (CFNS).Hum Mutat. 2005 Aug;26(2):113-8. doi: 10.1002/humu.20193. Hum Mutat. 2005. PMID: 15959873
-
Aberrant cell segregation in the craniofacial primordium and the emergence of facial dysmorphology in craniofrontonasal syndrome.PLoS Genet. 2020 Feb 24;16(2):e1008300. doi: 10.1371/journal.pgen.1008300. eCollection 2020 Feb. PLoS Genet. 2020. PMID: 32092051 Free PMC article.
-
A Family with Craniofrontonasal Syndrome: The First Report of Familial Cases of Craniofrontonasal Syndrome with Bilateral Cleft Lip and Palate.Cleft Palate Craniofac J. 2018 Aug;55(7):1026-1029. doi: 10.1597/15-347. Epub 2018 Feb 26. Cleft Palate Craniofac J. 2018. PMID: 28140668
-
Clinical and genetic aspects of craniofrontonasal syndrome: towards resolving a genetic paradox.Mol Genet Metab. 2005 Sep-Oct;86(1-2):110-6. doi: 10.1016/j.ymgme.2005.07.017. Mol Genet Metab. 2005. PMID: 16143553 Review.
-
Diverse clinical and genetic aspects of craniofrontonasal syndrome.Pediatr Neurol. 2011 Feb;44(2):83-7. doi: 10.1016/j.pediatrneurol.2010.10.012. Pediatr Neurol. 2011. PMID: 21215906 Review.
Cited by
-
Bone cell interactions through Eph/ephrin: bone modeling, remodeling and associated diseases.Cell Adh Migr. 2012 Mar-Apr;6(2):148-56. doi: 10.4161/cam.20888. Epub 2012 Mar 1. Cell Adh Migr. 2012. PMID: 22660185 Free PMC article. Review.
-
Neuronal guidance genes in health and diseases.Protein Cell. 2023 Apr 21;14(4):238-261. doi: 10.1093/procel/pwac030. Protein Cell. 2023. PMID: 36942388 Free PMC article. Review.
-
Presenilin-dependent intramembrane cleavage of ephrin-B1.Mol Neurodegener. 2006 Jun 12;1:2. doi: 10.1186/1750-1326-1-2. Mol Neurodegener. 2006. PMID: 16930449 Free PMC article.
-
Mutational analyses of UPIIIA, SHH, EFNB2 and HNF1beta in persistent cloaca and associated kidney malformations.J Pediatr Urol. 2007 Feb;3(1):2-9. doi: 10.1016/j.jpurol.2006.03.002. J Pediatr Urol. 2007. PMID: 17476318 Free PMC article.
-
Molecular Mechanisms Involved in Craniosynostosis.In Vivo. 2023 Jan-Feb;37(1):36-46. doi: 10.21873/invivo.13052. In Vivo. 2023. PMID: 36593018 Free PMC article. Review.
References
-
- Cohen, M. M., Jr. (1979) Birth Defects Orig. Artic. Ser. 15, 85–89. - PubMed
-
- Slover, R. & Sujansky, E. (1979) Birth Defects Orig. Artic. Ser. 15, 75–83. - PubMed
-
- Reynolds, J. F., Haas, R. J., Edgerton, M. T. & Kelly, T. E. (1982) J. Craniofac. Genet. Dev. Biol. 2, 233–238. - PubMed
-
- Sax, C. M. & Flannery, D. B. (1986) Clin. Genet. 29, 508–515. - PubMed
-
- Morris, C. A., Palumbos, J. C. & Carey, J. C. (1987) Am. J. Med. Genet. 27, 623–631. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
