

NSD1 mutations are the major cause of Sotos syndrome and occur in some cases of Weaver syndrome but are rare in other overgrowth phenotypes
- PMID: 12464997
- PMCID: PMC378618
- DOI: 10.1086/345647
NSD1 mutations are the major cause of Sotos syndrome and occur in some cases of Weaver syndrome but are rare in other overgrowth phenotypes
Abstract
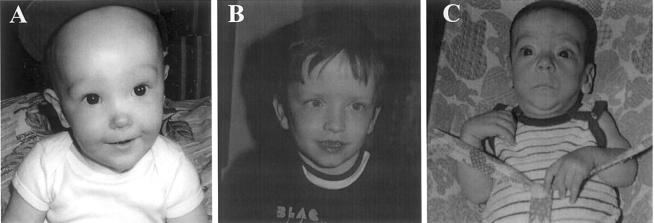
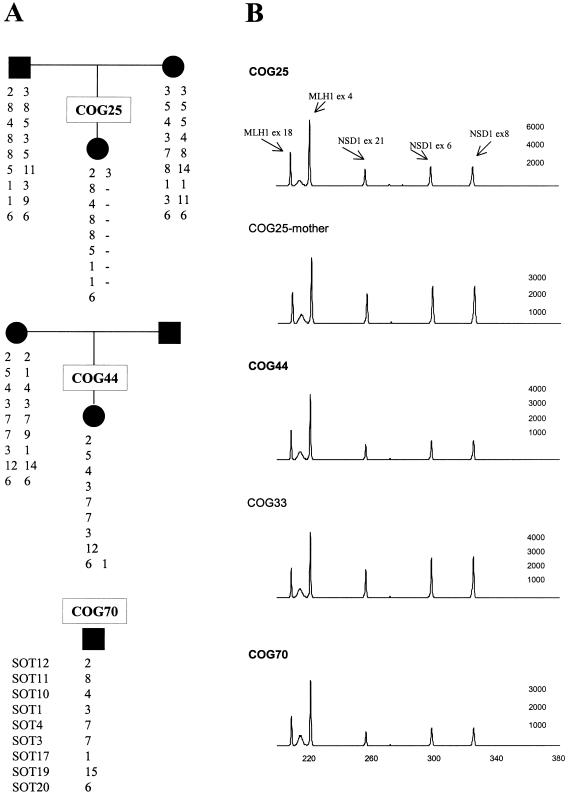
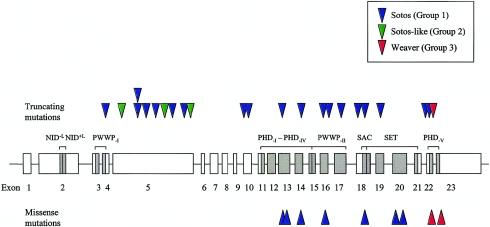
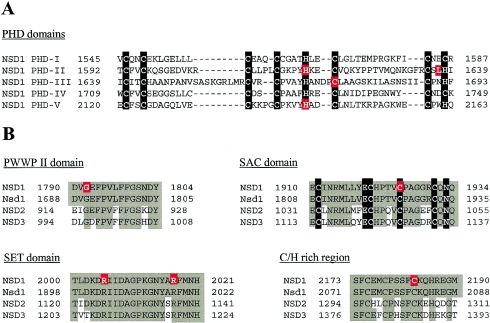
Sotos syndrome is a childhood overgrowth syndrome characterized by a distinctive facial appearance, height and head circumference >97th percentile, advanced bone age, and developmental delay. Weaver syndrome is characterized by the same criteria but has its own distinctive facial gestalt. Recently, a 2.2-Mb chromosome 5q35 microdeletion, encompassing NSD1, was reported as the major cause of Sotos syndrome, with intragenic NSD1 mutations identified in a minority of cases. We evaluated 75 patients with childhood overgrowth, for intragenic mutations and large deletions of NSD1. The series was phenotypically scored into four groups, prior to the molecular analyses: the phenotype in group 1 (n=37) was typical of Sotos syndrome; the phenotype in group 2 (n=13) was Sotos-like but with some atypical features; patients in group 3 (n=7) had Weaver syndrome, and patients in group 4 (n=18) had an overgrowth condition that was neither Sotos nor Weaver syndrome. We detected three deletions and 32 mutations (13 frameshift, 8 nonsense, 2 splice-site, and 9 missense) that are likely to impair NSD1 functions. The truncating mutations were spread throughout NSD1, but there was evidence of clustering of missense mutations in highly conserved functional domains between exons 13 and 23. There was a strong correlation between presence of an NSD1 alteration and clinical phenotype, in that 28 of 37 (76%) patients in group 1 had NSD1 mutations or deletions, whereas none of the patients in group 4 had abnormalities of NSD1. Three patients with Weaver syndrome had NSD1 mutations, all between amino acids 2142 and 2184. We conclude that intragenic mutations of NSD1 are the major cause of Sotos syndrome and account for some Weaver syndrome cases but rarely occur in other childhood overgrowth phenotypes.
Figures
Similar articles
-
Spectrum of NSD1 mutations in Sotos and Weaver syndromes.J Med Genet. 2003 Jun;40(6):436-40. doi: 10.1136/jmg.40.6.436. J Med Genet. 2003. PMID: 12807965 Free PMC article.
-
Mutations in NSD1 are responsible for Sotos syndrome, but are not a frequent finding in other overgrowth phenotypes.Eur J Hum Genet. 2003 Nov;11(11):858-65. doi: 10.1038/sj.ejhg.5201050. Eur J Hum Genet. 2003. PMID: 14571271
-
Genotype-phenotype associations in Sotos syndrome: an analysis of 266 individuals with NSD1 aberrations.Am J Hum Genet. 2005 Aug;77(2):193-204. doi: 10.1086/432082. Epub 2005 Jun 7. Am J Hum Genet. 2005. PMID: 15942875 Free PMC article.
-
NSD1 mutations in Sotos syndrome.Am J Med Genet C Semin Med Genet. 2005 Aug 15;137C(1):24-31. doi: 10.1002/ajmg.c.30061. Am J Med Genet C Semin Med Genet. 2005. PMID: 16010675 Review.
-
Molecular basis of Sotos syndrome.Horm Res. 2004;62 Suppl 3:60-5. doi: 10.1159/000080501. Horm Res. 2004. PMID: 15539801 Review.
Cited by
-
Identification of a 3.0-kb major recombination hotspot in patients with Sotos syndrome who carry a common 1.9-Mb microdeletion.Am J Hum Genet. 2005 Jan;76(1):52-67. doi: 10.1086/426950. Epub 2004 Nov 16. Am J Hum Genet. 2005. PMID: 15580547 Free PMC article.
-
Roles and regulation of histone methylation in animal development.Nat Rev Mol Cell Biol. 2019 Oct;20(10):625-641. doi: 10.1038/s41580-019-0151-1. Epub 2019 Jul 2. Nat Rev Mol Cell Biol. 2019. PMID: 31267065 Free PMC article. Review.
-
DMRscaler: a scale-aware method to identify regions of differential DNA methylation spanning basepair to multi-megabase features.BMC Bioinformatics. 2022 Sep 5;23(1):364. doi: 10.1186/s12859-022-04899-1. BMC Bioinformatics. 2022. PMID: 36064314 Free PMC article.
-
Molecular and comparative genetics of mental retardation.Genetics. 2004 Feb;166(2):835-81. doi: 10.1534/genetics.166.2.835. Genetics. 2004. PMID: 15020472 Free PMC article.
-
An inactivating mutation in the histone deacetylase SIRT6 causes human perinatal lethality.Genes Dev. 2018 Mar 1;32(5-6):373-388. doi: 10.1101/gad.307330.117. Epub 2018 Mar 19. Genes Dev. 2018. PMID: 29555651 Free PMC article.
References
Electronic-Database Information
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for the NSD1 genomic sequence [accession number AF395588])
-
- NCBI BLAST Home Page, http://www.ncbi.nlm.nih.gov/blast/ (for comparison of human NSD1 [GenBank accession number AF395588; RefSeq protein sequence NP_071900] with mouse Nsd1 [GenBank accession number AF419220, RefSeq protein sequence NP_032765] and human NSD2 [GenBank accession number AF083389, RefSeq protein sequence NP_579890] and human NSD3 [GenBank accession number AF332469, RefSeq protein sequence NP_075447])
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for Sotos syndrome [MIM 117550], Weaver Syndrome [MIM 277590], Marshall Smith syndrome [MIM 602535], autosomal dominant macrocephaly [MIM 605309, MIM 153470], Beckwith-Wiedemann syndrome [MIM 130650], Simpson-Golabi Behmel syndrome [MIM 312870], Neurofibromatosis type 1 [MIM 162200], Williams syndrome [MIM 194050], Smith-Magenis syndrome [MIM 182290])
-
- Primer3, http://www-genome.wi.mit.edu/cgi-bin/primer/primer3_www.cgi (for designing microsatellite markers amplifying repetitive elements in the vicinity of NSD1 and NSD1 mutation screening primers)
-
- UCSC Genome Bioinformatics, http://genome.ucsc.edu/ (for identification and positioning of repetitive sequence elements flanking NSD1)
References
-
- Aasland R, Gibson TJ, Stewart AF (1995) The PHD finger: implications for chromatin-mediated transcriptional regulation. Trends Biochem Sci 20:56–59 - PubMed
-
- Angrand PO, Apiou F, Stewart AF, Dutrillaux B, Losson R, Chambon P (2001) NSD3, a new SET domain-containing gene, maps to 8p12 and is amplified in human breast cancer cell lines. Genomics 74:79–88 - PubMed
-
- Charbonnier F, Olschwang S, Wang Q, Boisson C, Martin C, Buisine M-P, Puisieux A, Frebourg T (2002) MSH2 in contrast to MLH1 and MSH6 is frequently inactivated by exonic and promoter rearrangements in hereditary nonpolyposis colorectal cancer. Cancer Res 62:848–853 - PubMed
-
- Chesi M, Nardini E, Lim RS, Smith KD, Kuehl WM, Bergsagel PL (1998) The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood 92:3025–3034 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
