

Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes
- PMID: 12219090
- PMCID: PMC3169431
- DOI: 10.1038/ng985
Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes
Abstract
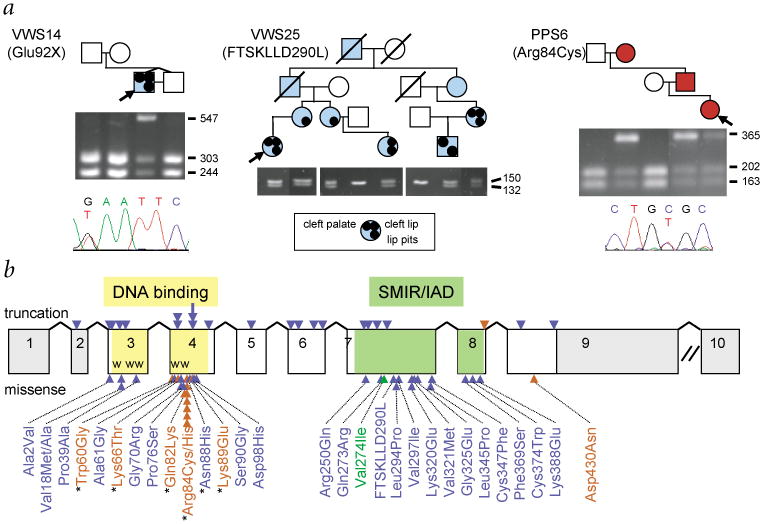
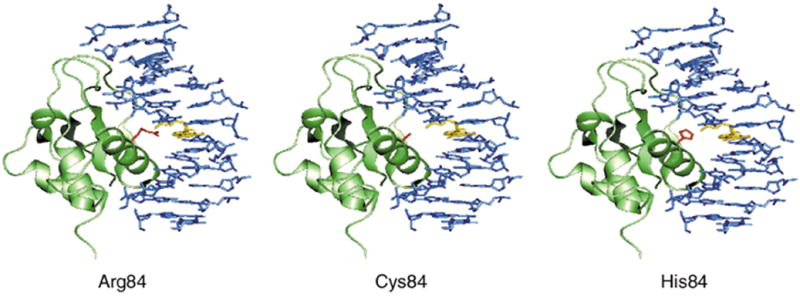
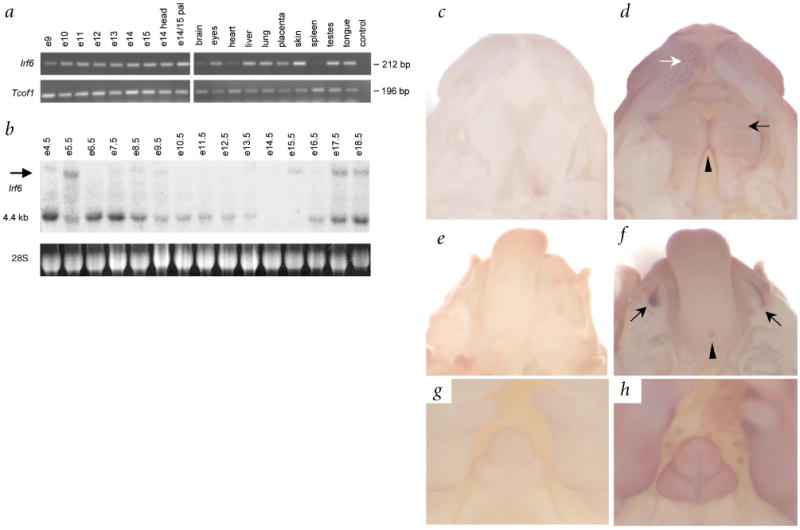
Interferon regulatory factor 6 (IRF6) belongs to a family of nine transcription factors that share a highly conserved helix-turn-helix DNA-binding domain and a less conserved protein-binding domain. Most IRFs regulate the expression of interferon-alpha and -beta after viral infection, but the function of IRF6 is unknown. The gene encoding IRF6 is located in the critical region for the Van der Woude syndrome (VWS; OMIM 119300) locus at chromosome 1q32-q41 (refs 2,3). The disorder is an autosomal dominant form of cleft lip and palate with lip pits, and is the most common syndromic form of cleft lip or palate. Popliteal pterygium syndrome (PPS; OMIM 119500) is a disorder with a similar orofacial phenotype that also includes skin and genital anomalies. Phenotypic overlap and linkage data suggest that these two disorders are allelic. We found a nonsense mutation in IRF6 in the affected twin of a pair of monozygotic twins who were discordant for VWS. Subsequently, we identified mutations in IRF6 in 45 additional unrelated families affected with VWS and distinct mutations in 13 families affected with PPS. Expression analyses showed high levels of Irf6 mRNA along the medial edge of the fusing palate, tooth buds, hair follicles, genitalia and skin. Our observations demonstrate that haploinsufficiency of IRF6 disrupts orofacial development and are consistent with dominant-negative mutations disturbing development of the skin and genitalia.
Figures
Comment in
-
The pit, the cleft and the web.Nat Genet. 2002 Oct;32(2):219-20. doi: 10.1038/ng1002-219. Nat Genet. 2002. PMID: 12355077 No abstract available.
Similar articles
-
Novel mutations in the IRF6 gene for Van der Woude syndrome.Hum Genet. 2003 Oct;113(5):382-6. doi: 10.1007/s00439-003-0989-2. Epub 2003 Aug 14. Hum Genet. 2003. PMID: 12920575
-
Genomic, cDNA and embryonic expression analysis of zebrafish IRF6, the gene mutated in the human oral clefting disorders Van der Woude and popliteal pterygium syndromes.Gene Expr Patterns. 2005 Jun;5(5):629-38. doi: 10.1016/j.modgep.2005.03.002. Epub 2005 Apr 19. Gene Expr Patterns. 2005. PMID: 15939375
-
Missense mutations that cause Van der Woude syndrome and popliteal pterygium syndrome affect the DNA-binding and transcriptional activation functions of IRF6.Hum Mol Genet. 2009 Feb 1;18(3):535-45. doi: 10.1093/hmg/ddn381. Epub 2008 Nov 26. Hum Mol Genet. 2009. PMID: 19036739 Free PMC article.
-
Non-random distribution of deleterious mutations in the DNA and protein-binding domains of IRF6 are associated with Van Der Woude syndrome.Mol Genet Genomic Med. 2020 Aug;8(8):e1355. doi: 10.1002/mgg3.1355. Epub 2020 Jun 17. Mol Genet Genomic Med. 2020. PMID: 32558391 Free PMC article. Review.
-
Toward an orofacial gene regulatory network.Dev Dyn. 2016 Mar;245(3):220-32. doi: 10.1002/dvdy.24341. Epub 2015 Sep 17. Dev Dyn. 2016. PMID: 26332872 Free PMC article. Review.
Cited by
-
A multi-ethnic genome-wide association study identifies novel loci for non-syndromic cleft lip with or without cleft palate on 2p24.2, 17q23 and 19q13.Hum Mol Genet. 2016 Jul 1;25(13):2862-2872. doi: 10.1093/hmg/ddw104. Epub 2016 Mar 30. Hum Mol Genet. 2016. PMID: 27033726 Free PMC article.
-
Plasticity-induced repression of Irf6 underlies acquired resistance to cancer immunotherapy in pancreatic ductal adenocarcinoma.Nat Commun. 2024 Feb 20;15(1):1532. doi: 10.1038/s41467-024-46048-7. Nat Commun. 2024. PMID: 38378697 Free PMC article.
-
Susceptibility to DNA damage as a molecular mechanism for non-syndromic cleft lip and palate.PLoS One. 2013 Jun 12;8(6):e65677. doi: 10.1371/journal.pone.0065677. Print 2013. PLoS One. 2013. PMID: 23776525 Free PMC article.
-
IRF6 is the mediator of TGFβ3 during regulation of the epithelial mesenchymal transition and palatal fusion.Sci Rep. 2015 Aug 4;5:12791. doi: 10.1038/srep12791. Sci Rep. 2015. PMID: 26240017 Free PMC article.
-
De novo 2.3 Mb microdeletion of 1q32.2 involving the Van der Woude Syndrome locus.Mol Cytogenet. 2013 Aug 6;6:31. doi: 10.1186/1755-8166-6-31. eCollection 2013. Mol Cytogenet. 2013. PMID: 23915469 Free PMC article.
References
-
- Taniguchi T, Ogasawara K, Takaoka A, Tanaka N. IRF family of transcription factors as regulators of host defense. Annu Rev Immunol. 2001;19:623–655. - PubMed
-
- Gorlin RJ, Sedano HO, Cervenka J. Popliteal pterygium syndrome. A syndrome comprising cleft lip-palate, popliteal and intercrural pterygia, digital and genital anomalies. Pediatrics. 1968;41:503–509. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P60 DE013076-01S10005/DE/NIDCR NIH HHS/United States
- R01 DE014667/DE/NIDCR NIH HHS/United States
- R01 DE014667-03/DE/NIDCR NIH HHS/United States
- P60 DE013076-010005/DE/NIDCR NIH HHS/United States
- P60 DE013076-020005/DE/NIDCR NIH HHS/United States
- P60 DE013076-040005/DE/NIDCR NIH HHS/United States
- R01 DE014667-04/DE/NIDCR NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- P60 DE013076/DE/NIDCR NIH HHS/United States
- R01 DE014667-01/DE/NIDCR NIH HHS/United States
- R01 DE014667-02/DE/NIDCR NIH HHS/United States
- P60 DE013076-03S10005/DE/NIDCR NIH HHS/United States
- P60 DE013076-030005/DE/NIDCR NIH HHS/United States
- P60 DE013076-050005/DE/NIDCR NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
