

Functional analysis of RUNX2 mutations in Japanese patients with cleidocranial dysplasia demonstrates novel genotype-phenotype correlations
- PMID: 12196916
- PMCID: PMC378531
- DOI: 10.1086/342717
Functional analysis of RUNX2 mutations in Japanese patients with cleidocranial dysplasia demonstrates novel genotype-phenotype correlations
Abstract
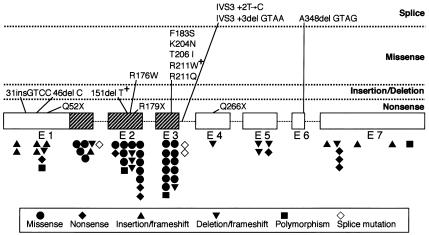
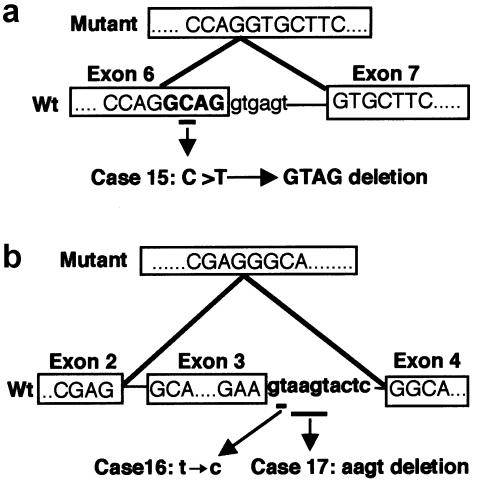
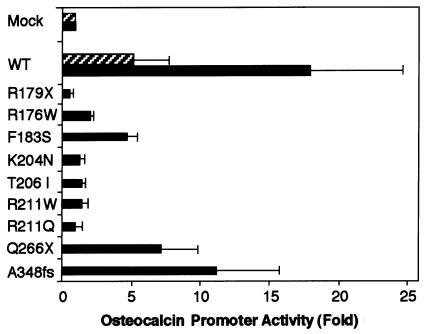
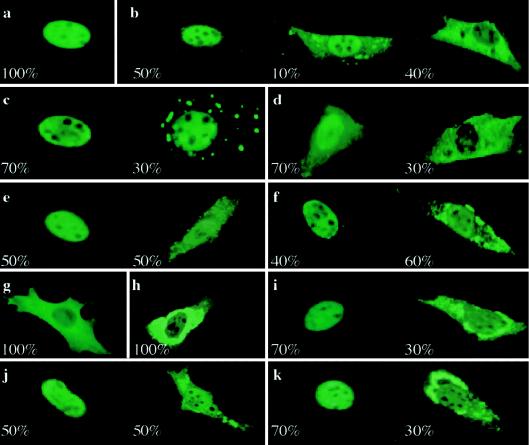
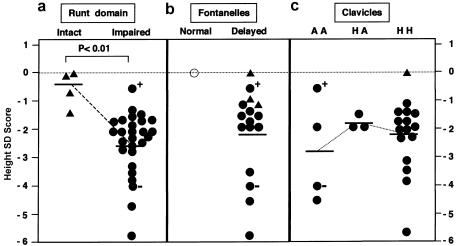
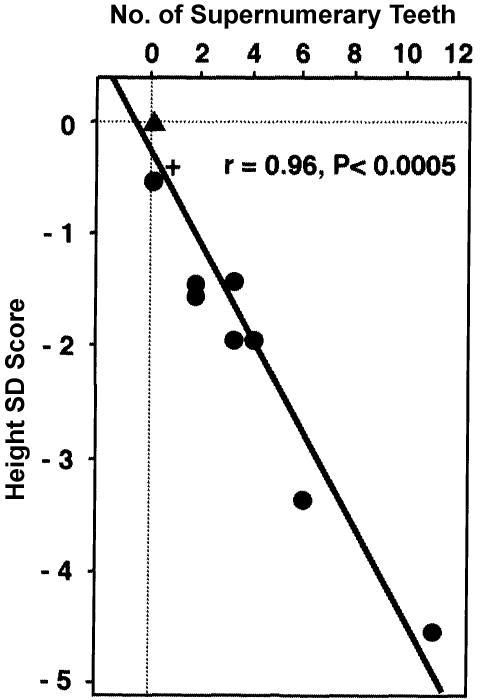
Cleidocranial dysplasia (CCD) is an autosomal dominant heritable skeletal disease caused by heterozygous mutations in the osteoblast-specific transcription factor RUNX2. We have performed mutational analysis of RUNX2 on 24 unrelated patients with CCD. In 17 patients, 16 distinct mutations were detected in the coding region of RUNX2: 4 frameshift, 3 nonsense, 6 missense, and 2 splicing mutations, in addition to 1 polymorphism. The missense mutations were all clustered within the Runt domain, and their protein products were severely impaired in DNA binding and transactivation. In contrast, two RUNX2 mutants had the Runt domain intact and remained partially competent for transactivation. One criterion of CCD, short stature, was much milder in the patients with the intact Runt domain than in those without. Furthermore, a significant correlation was found between short stature and the number of supernumerary teeth. On the one hand, these genotype-phenotype correlations highlight a general, quantitative dependency, by skeleto-dental developments, on the gene dosage of RUNX2, which has hitherto been obscured by extreme clinical diversities of CCD; this gene-dosage effect is presumed to manifest on small reductions in the total RUNX2 activity, by approximately one-fourth of the normal level at minimum. On the other hand, the classic CCD phenotype, hypoplastic clavicles or open fontanelles, was invariably observed in all patients, including those with normal height. Thus, the cleidocranial bone formation, as mediated by intramembranous ossification, may require a higher level of RUNX2 than does skeletogenesis (mediated by endochondral ossification), as well as odontogenesis (involving still different complex processes). Overall, these results suggest that CCD could result from much smaller losses in the RUNX2 function than has been envisioned on the basis of the conventional haploinsufficiency model.
Figures



Similar articles
-
Functional analysis of RUNX2 mutations in cleidocranial dysplasia: novel insights into genotype-phenotype correlations.Blood Cells Mol Dis. 2003 Mar-Apr;30(2):184-93. doi: 10.1016/s1079-9796(03)00020-2. Blood Cells Mol Dis. 2003. PMID: 12732182
-
Cleidocranial dysplasia: oral features and genetic analysis of 11 patients.Oral Dis. 2012 Mar;18(2):184-90. doi: 10.1111/j.1601-0825.2011.01862.x. Epub 2011 Oct 24. Oral Dis. 2012. PMID: 22023169
-
Mutations in the RUNX2 gene in patients with cleidocranial dysplasia.Hum Mutat. 2002 Mar;19(3):209-16. doi: 10.1002/humu.10043. Hum Mutat. 2002. PMID: 11857736 Review.
-
Identification of a novel frameshift mutation (383insT) in the RUNX2 (PEBP2 alpha/CBFA1/AML3) gene in a Japanese patient with cleidocranial dysplasia.J Bone Miner Metab. 2001;19(4):263-6. doi: 10.1007/s007740170030. J Bone Miner Metab. 2001. PMID: 11448020
-
Cleidocranial dysplasia with severe parietal bone dysplasia: C-terminal RUNX2 mutations.Birth Defects Res A Clin Mol Teratol. 2006 Feb;76(2):78-85. doi: 10.1002/bdra.20231. Birth Defects Res A Clin Mol Teratol. 2006. PMID: 16463420 Review.
Cited by
-
Aurora kinase-induced phosphorylation excludes transcription factor RUNX from the chromatin to facilitate proper mitotic progression.Proc Natl Acad Sci U S A. 2016 Jun 7;113(23):6490-5. doi: 10.1073/pnas.1523157113. Epub 2016 May 23. Proc Natl Acad Sci U S A. 2016. PMID: 27217562 Free PMC article.
-
Disease mutations in RUNX1 and RUNX2 create nonfunctional, dominant-negative, or hypomorphic alleles.EMBO J. 2007 Feb 21;26(4):1163-75. doi: 10.1038/sj.emboj.7601568. Epub 2007 Feb 8. EMBO J. 2007. PMID: 17290219 Free PMC article.
-
Cleidocranial dysplasia with growth hormone deficiency: a case report.BMC Pediatr. 2020 Jan 16;20(1):19. doi: 10.1186/s12887-020-1914-8. BMC Pediatr. 2020. PMID: 31948427 Free PMC article.
-
Cleidocranial dysplasia: a case report.J Clin Res Pediatr Endocrinol. 2010;2(3):134-6. doi: 10.4274/jcrpe.v2i3.134. Epub 2010 Aug 9. J Clin Res Pediatr Endocrinol. 2010. PMID: 21274329 Free PMC article.
-
Manifestation and treatment in a cleidocranial dysplasia patient with a RUNX2 (T420I) mutation.Maxillofac Plast Reconstr Surg. 2015 Nov 12;37(1):41. doi: 10.1186/s40902-015-0042-0. eCollection 2015 Dec. Maxillofac Plast Reconstr Surg. 2015. PMID: 26594640 Free PMC article.
References
Electronic-Database Information
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for RUNX2 [accession number AF001450])
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for CCD [MIM 119600])
References
-
- Bae SC, Takahashi E, Zhang YW, Ogawa E, Shigesada K, Namba Y, Satake M, Ito Y (1995) Cloning, mapping and expression of PEBP2αC, a third gene encoding the mammalian Runt domain. Gene 159:245–248 - PubMed
-
- Bae SC, Yamaguchi-Iwai Y, Ogawa E, Maruyama M, Inuzuka M, Kagoshima H, Shigesada K, Satake M, Ito Y (1993) Isolation of PEBP2αB cDNA representing the mouse homolog of human acute myeloid leukemia gene, AML1. Oncogene 8:809–814 - PubMed
-
- Bravo J, Li Z, Speck NA, Warren AJ (2001) The leukemia-associated AML1 (Runx1)–CBFβ complex functions as a DNA-induced molecular clamp. Nat Struct Biol 8:371–378 - PubMed
-
- Chitayat D, Hodgkinson KA, Azouz EM (1992) Intrafamilial variability in cleidocranial dysplasia: a three generation family. Am J Med Genet 42:298–303 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
