

A 1.5 million-base pair inversion polymorphism in families with Williams-Beuren syndrome
- PMID: 11685205
- PMCID: PMC2889916
- DOI: 10.1038/ng753
A 1.5 million-base pair inversion polymorphism in families with Williams-Beuren syndrome
Abstract
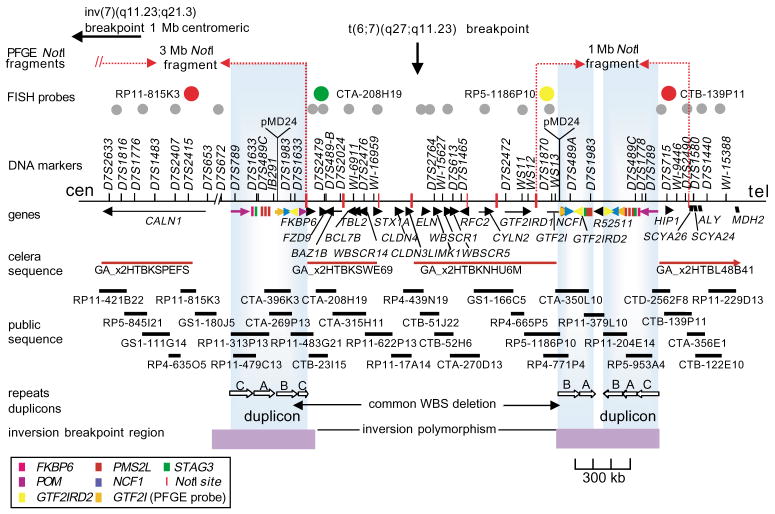
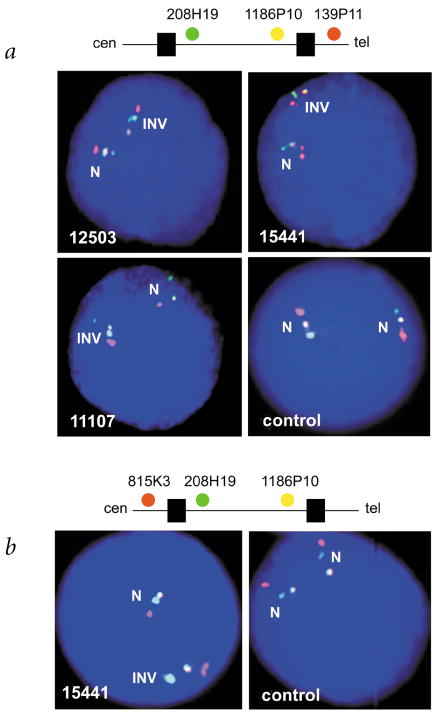
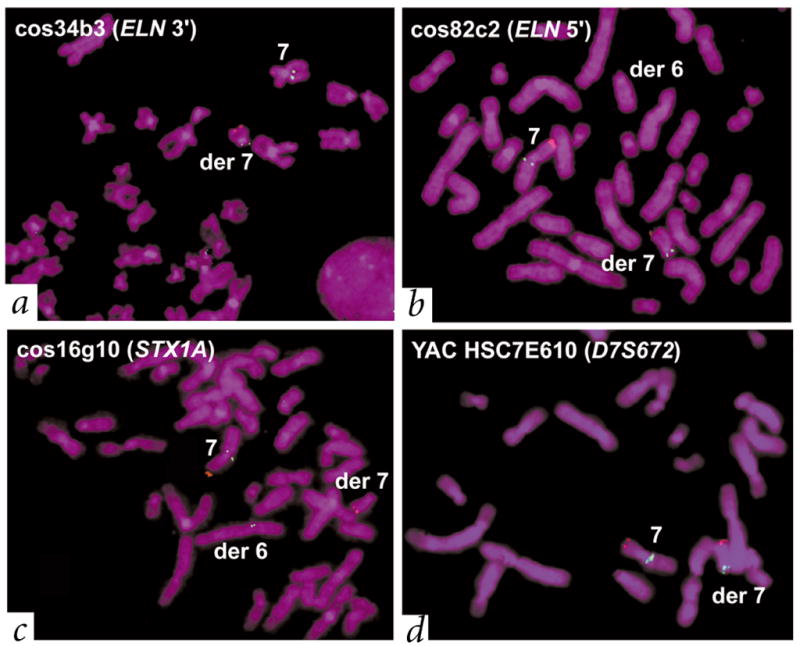
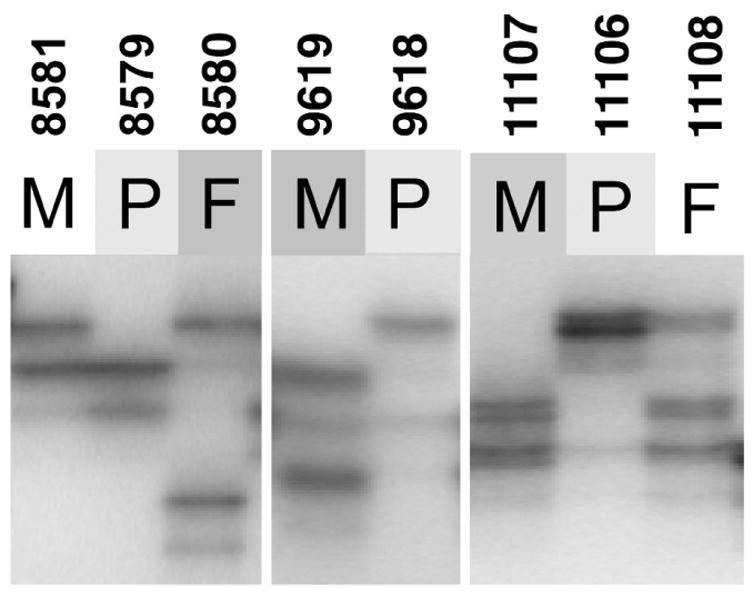
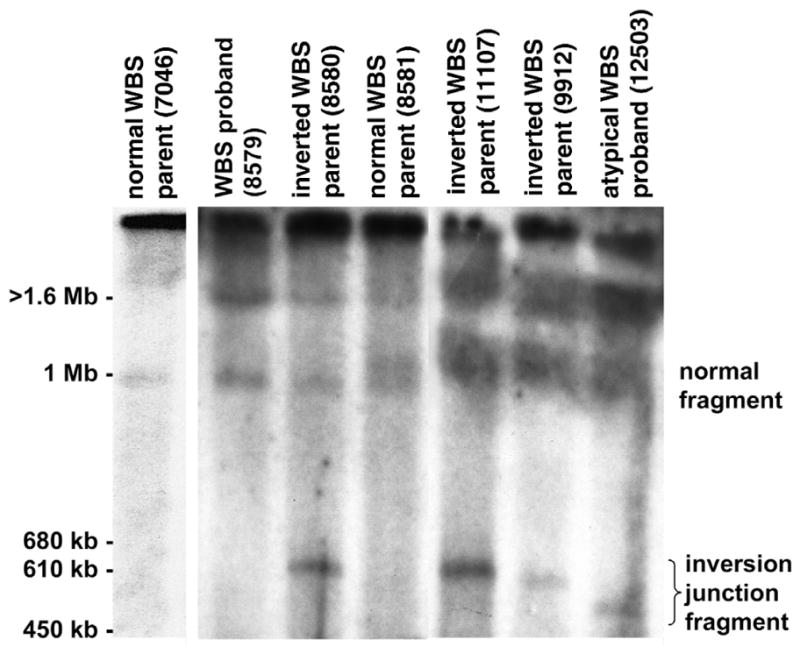
Williams-Beuren syndrome (WBS) is most often caused by hemizygous deletion of a 1.5-Mb interval encompassing at least 17 genes at 7q11.23 (refs. 1,2). As with many other haploinsufficiency diseases, the mechanism underlying the WBS deletion is thought to be unequal meiotic recombination, probably mediated by the highly homologous DNA that flanks the commonly deleted region. Here, we report the use of interphase fluorescence in situ hybridization (FISH) and pulsed-field gel electrophoresis (PFGE) to identify a genomic polymorphism in families with WBS, consisting of an inversion of the WBS region. We have observed that the inversion is hemizygous in 3 of 11 (27%) atypical affected individuals who show a subset of the WBS phenotypic spectrum but do not carry the typical WBS microdeletion. Two of these individuals also have a parent who carries the inversion. In addition, in 4 of 12 (33%) families with a proband carrying the WBS deletion, we observed the inversion exclusively in the parent transmitting the disease-related chromosome. These results suggest the presence of a newly identified genomic variant within the population that may be associated with the disease. It may result in predisposition to primarily WBS-causing microdeletions, but may also cause translocations and inversions.
Figures
Similar articles
-
Observation of a parental inversion variant in a rare Williams-Beuren syndrome family with two affected children.Hum Genet. 2005 Aug;117(4):383-8. doi: 10.1007/s00439-005-1325-9. Epub 2005 Jun 3. Hum Genet. 2005. PMID: 15933846 Free PMC article.
-
Fine-scale comparative mapping of the human 7q11.23 region and the orthologous region on mouse chromosome 5G: the low-copy repeats that flank the Williams-Beuren syndrome deletion arose at breakpoint sites of an evolutionary inversion(s).Genomics. 2000 Oct 1;69(1):1-13. doi: 10.1006/geno.2000.6312. Genomics. 2000. PMID: 11013070
-
Copy number variation at the 7q11.23 segmental duplications is a susceptibility factor for the Williams-Beuren syndrome deletion.Genome Res. 2008 May;18(5):683-94. doi: 10.1101/gr.073197.107. Epub 2008 Feb 21. Genome Res. 2008. PMID: 18292220 Free PMC article.
-
The genomic basis of the Williams-Beuren syndrome.Cell Mol Life Sci. 2009 Apr;66(7):1178-97. doi: 10.1007/s00018-008-8401-y. Cell Mol Life Sci. 2009. PMID: 19039520 Free PMC article. Review.
-
Familial Williams-Beuren syndrome showing varying clinical expression.Am J Med Genet. 2001 Feb 1;98(4):324-9. doi: 10.1002/1096-8628(20010201)98:4<324::aid-ajmg1103>3.0.co;2-5. Am J Med Genet. 2001. PMID: 11170076 Review.
Cited by
-
A role for transcription factor GTF2IRD2 in executive function in Williams-Beuren syndrome.PLoS One. 2012;7(10):e47457. doi: 10.1371/journal.pone.0047457. Epub 2012 Oct 31. PLoS One. 2012. PMID: 23118870 Free PMC article.
-
Williams-Beuren Syndrome: A Clinical Study of 55 Brazilian Patients and the Diagnostic Use of MLPA.Biomed Res Int. 2015;2015:903175. doi: 10.1155/2015/903175. Epub 2015 May 18. Biomed Res Int. 2015. PMID: 26090456 Free PMC article.
-
Wolf-Hirschhorn syndrome-associated chromosome changes are not mediated by olfactory receptor gene clusters nor by inversion polymorphism on 4p16.Hum Genet. 2007 Dec;122(5):423-30. doi: 10.1007/s00439-007-0412-5. Epub 2007 Aug 4. Hum Genet. 2007. PMID: 17676343
-
Inversion polymorphisms and non-contiguous terminal deletions: the cause and the (unpredicted) effect of our genome architecture.J Med Genet. 2006 May;43(5):e19. doi: 10.1136/jmg.2005.037671. J Med Genet. 2006. PMID: 16648372 Free PMC article.
-
Genomic inversions and GOLGA core duplicons underlie disease instability at the 15q25 locus.PLoS Genet. 2019 Mar 27;15(3):e1008075. doi: 10.1371/journal.pgen.1008075. eCollection 2019 Mar. PLoS Genet. 2019. PMID: 30917130 Free PMC article.
References
-
- Ewart AK, et al. Hemizygosity at the elastin locus in a developmental disorder, Williams syndrome. Nature Genet. 1993;5:11–16. - PubMed
-
- Osborne LR. Williams-Beuren syndrome—unraveling the mysteries of a microdeletion disorder. Mol Genet Metab. 1999;67:1–10. - PubMed
-
- Baumer A, et al. High level of unequal meiotic crossovers at the origin of the 22q11. 2 and 7q11.23 deletions. Hum Mol Genet. 1998;7:887–894. - PubMed
-
- Greenberg F. Williams syndrome professional symposium. Am J Med Genet Suppl. 1990;6:85–88. - PubMed
-
- Pober BR, Dykens EM. Williams syndrome: an overview of medical, cognitive, and behavioural features. Child Adoles Psychiatr Clin N Am. 1996;5:929–943.
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
